

薬生薬審発0202第1号(平成29年2月2日)
ICH HARMONISED GUIDELINE- M4E(R2) – Dated 15 June 2016
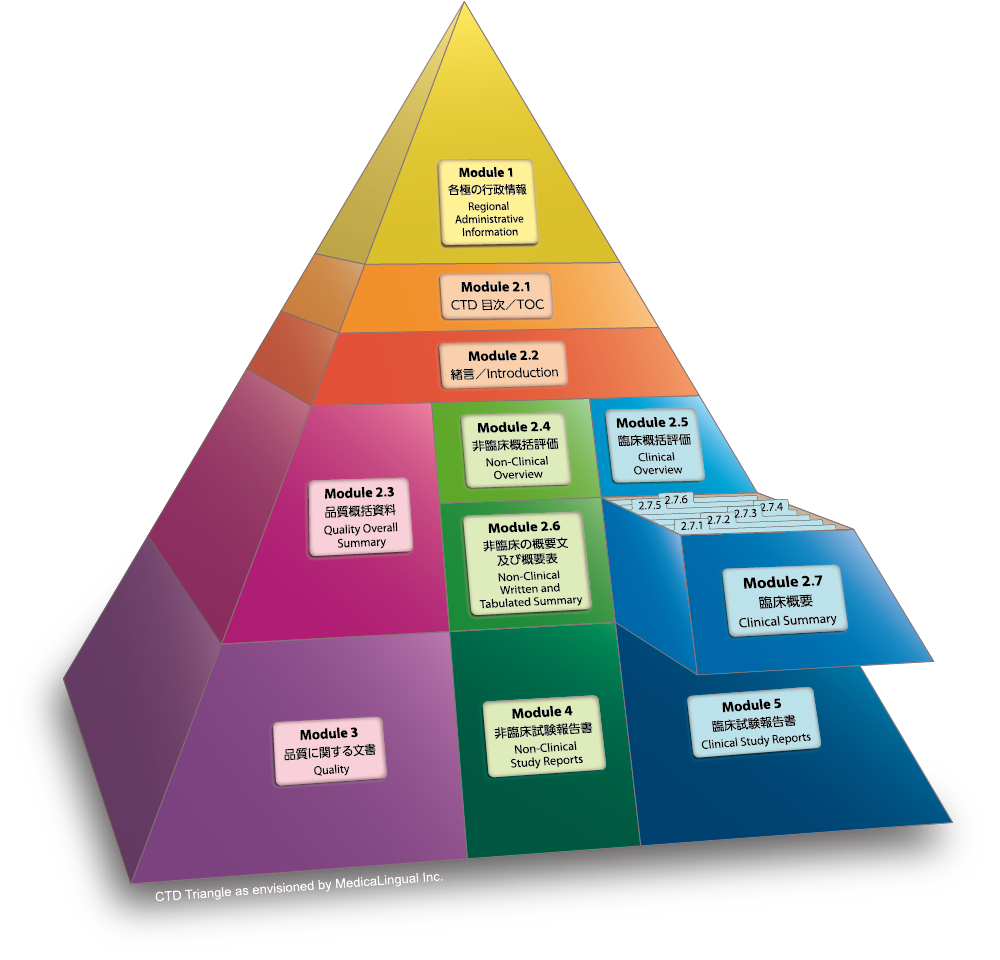
Module 2.7 臨床概要
序文
臨床概要の目的は、国際共通化資料(CTD)中の全ての臨床情報の詳細な事実に基づく要約を提供することである。これには、ICH E3 でいう「治験の総括報告書」から得られた情報、第 5 部に報告書一式が添付されている試験のメタアナリシスや複数試験にまたがる併合解析から得られた情報、他の地域で販売されている品目については市販後のデータが含まれる。本文書で提供される試験間の結果の比較及び解析は、事実に基づく観察・結果に重点を置くこと。対照的に、「臨床に関する概括評価」では、臨床において得られた情報についての考察と解釈、及び既存の治療法の中における当該医薬品の位置付けに関する考察を含め、臨床試験計画とその結果に関する重要な解 析を示すこと。
臨床概要の長さは伝達すべき情報により変わるが、付表を除き通常 50 ページから 400 ページの範囲と想定されている。
Module 2.7 CLINICAL SUMMARY
Preamble
The Clinical Summary is intended to provide a detailed, factual summarisation of all of the clinical information in the Common Technical Document. This includes information provided in ICH E3 clinical study reports; information obtained from any meta-analyses or other cross-study analyses for which full reports have been included in Module 5; and postmarketing data for products that have been marketed in other regions. The comparisons and analyses of results across studies provided in this document should focus on factual observations. In contrast, the CTD Clinical Overview document should provide critical analysis of the clinical study program and its results, including discussion and interpretation of the clinical findings and discussion of the place of the test drug in the armamentarium.
The length of the Clinical Summary will vary substantially according to the information to be conveyed, but it is anticipated that (excluding attached tables) the Clinical Summary will usually be in the range of 50 to 400 pages.
2.7.1 生物薬剤学及び関連する分析法の概要
2.7.1.1 背景及び概観
本項では製剤開発過程、in vitro 及び in vivo での製剤特性、バイオアベイラビリティ(BA)、比較 BA、生物学的同等性(BE)、in vitro 溶出特性に関する情報の収集に用いた方法と根拠の概観を示すこと。試験の計画及び実施に際して参照したガイドライン又は文献を示すこと。また、本項では用いた分析法の概観について、特に分析法バリデーションの成績特性(例:線形範囲、感度、特異性)及び品質管理(例:正確性及び精度)に重点をおいて示すこと。本項には個々の試験に関する詳細な情報は記載しないこと。
2.7.1 Summary of Biopharmaceutic Studies and Associated Analytical Methods
2.7.1.1 Background and Overview
This section should provide the reviewer with an overall view of the formulation development process, the in vitro and in vivo dosage form performance, and the general approach and rationale used in developing the bioavailability (BA), comparative BA, bioequivalence (BE), and in vitro dissolution profile database. Reference should be made to any guidelines or literature used in planning and conducting the studies. This section should also provide the reviewer with an overview of the analytical methods used, with emphasis on the performance characteristics of assay validation (e.g., linearity range, sensitivity, specificity) and quality control (e.g., accuracy and precision). This section should not include detailed information about individual studies.
2.7.1.2 個々の試験結果の要約
全ての生物薬剤学試験を基本的には一覧表で提示し(第 2.7.1.4 項 付録を参照)、BA 及びBE に関する in vitro 又は in vivo の重要なデータと情報が得られた個々の試験の内容と結果について、簡単な説明を付け加えること。説明は、論文抄録のように簡潔なものとし、デザイン上の特徴及び重要な結果のみを記載すること。試験デザインが似ているものはまとめて表記してもよい。その場合、結果がどの試験のものかが分るように記載し、試験間の結果に重要な差異があれば示すこと。これらの記述は、総括報告書の概要(シノプシス)(ICH E3)から抜粋してもよい。また、説明文中に、本項から各総括報告書へ参照できるように配慮しておくか、電子的リンクを貼りつけておくこと。
2.7.1.2 Summary of Results of Individual Studies
A tabular listing of all biopharmaceutic studies should generally be provided (see 2.7.1.4 Appendix), together with narrative descriptions of relevant features and outcomes of each of the individual studies that provided important in vitro or in vivo data and information relevant to BA and BE. The narrative descriptions should be brief, e.g., similar to an abstract for a journal article, and should describe critical design features and critical results. Similar studies may be described together, noting the individual study results and any important differences among the studies. These narratives may be abstracted from the ICH E3 synopsis. References or electronic links to the full report of each study should be included in the narratives.
2.7.1.3 全試験を通しての結果の比較と解析
本項では、原薬及び製剤について実施された全ての in vitro 溶出試験、BA 試験及び比較BA 試験についての事実に基づく要約を、特に試験間の結果の違いに注目して示すこと。この概観では、以下のことを考慮して、一般的に本文及び表(第 2.7.1.4 項 付録を参照)を用いて結果を要約すること。
- 製剤処方や製造法の変更が、in vitro の溶出及び BA、そして BE に関する結論に及ぼす影響についてのデータ。複雑な構造を持つ有効成分(例:タンパク質)を含む医薬品について製造法又は製剤処方の変更を行う場合は、変更前後の医薬品を比較する薬物動態(PK)試験を行い、変更の結果として PK 特性が変化しないことを確認すること。こうした試験は、しばしば BE 試験と呼ばれるものの、一般的には製剤からの有効成分の放出を評価することには焦点をあてていない。しかし、このような試験は本項で報告すること。また、PK 試験だけでは製剤間の類似性を確認するには必ずしも十分でないことに注意すること。多くの場合に、薬力学(PD)試験又は臨床試験が必要となり、状況によっては、さらに抗原性データも必要となることがあるかもしれない。これらの試験が実施された場合、その結果を申請資料の適当な場所に添付すること。
- 食事の種類又は食事のタイミングについて、BA 及び BE 判定に対する食事の影響の程度に関するデータ(適切な場合)。
- 溶出に対する pH の影響を含む in vitro 溶出性と BA との相関性に関するデータ及び溶出規格に関する結論。
- 異なる含量の製剤について BE 判定を含む比較 BA。
- 有効性を検証した臨床試験に用いた製剤と市販用製剤との比較 BA。
- 比較 BA試験において各製剤で認められた被験者間変動及び被験者内変動の原因と大きさ。
2.7.1.3 Comparison and Analyses of Results Acsross Studies
This section should provide a factual summary of all in vitro dissolution, BA, and comparative BA studies carried out with the drug substance or drug product, with particular attention to differences in results across studies. This overview should typically summarise the findings in text and tables (see 2.7.1.4 Appendix) and should consider the following:
- evidence of the effects of formulation and manufacturing changes on in vitro dissolution and BA and conclusions regarding BE. When manufacturing or formulation changes are made for products containing complex drug substances (e.g., a protein), pharmacokinetic (PK) studies comparing the product before and after the changes may be performed to ensure that the PK characteristics have not changed as a result of product changes. Although such studies are sometimes referred to as BE studies, they generally do not focus on assessing release of drug substance from drug product. Nonetheless, such studies should be reported in this section. Note also that PK studies alone may not be sufficient to assure similarity between such drug products. In many situations, pharmacodynamic (PD) studies or clinical trials may be necessary. Additionally, depending on the circumstances, antigenicity data may also be needed. Results of these other types of studies, when they are needed, should be reported in the appropriate places in the dossier.
- evidence of the extent of food effects on BA and conclusions regarding BE with respect to meal type or timing of the meal (where appropriate).
- evidence of correlations between in vitro dissolution and BA, including the effects of pH on dissolution, and conclusions regarding dissolution specifications.
- comparative bioavailability, including BE conclusions, for different dosage form strengths.
- comparative BA of the clinical study formulations (for clinical studies providing substantial evidence of efficacy) and the formulations to be marketed.
- the source and magnitude of observed inter- and intrasubject variability for each formulation in a comparative BA study.
2.7.1.4 付録
文書が読みやすくなるのであれば、図や表を適切に文章中に挿入すること。大きな表は、本項の最後に付録として添付することができる。
表 2.7.1.1 及び表 2.7.1.2 はそれぞれ BA 試験及び in vitro 溶出試験に関する情報や結果を報告するための表様式の例である。これらの表は、結果はもちろん試験の種類とデザインをも特定する。BE 試験の結果を報告するために作成された表には、Cmax 及び AUC の幾何平均値の比(試験製剤/対照製剤)並びにその 90%信頼区間、又は現在推奨されている BE 評価に対する基準も含める。
これらの表はテンプレートではなく、申請者が生物薬剤学試験をまとめる際に考慮すべき情報の種類を単に示したものである。これらの試験から得られた情報や結果が表、文章、図を用いて最も分かりやすく示されているかについても判断すること。例えば、結果が文章及び図で良く示されている場合は、表は試験の一覧を示すためだけに用いられることになる。
2.7.1.4 Appendix
Tables and figures should be embedded in the text of the appropriate sections when they enhance the readability of the document. Lengthy tables can be provided in the appendix at the end of the Section.
Tables 2.7.1.1 and 2.7.1.2 are provided as examples of tabular formats for reporting information and results related to bioavailability and in vitro dissolution studies respectively. These examples give results as well as identifying the type and design of the study. Tables prepared for reporting the results of BE studies could also include the mean ratios (test/reference) for Cmax and AUC and their 90% confidence interval, or the currently recommended metrics for BE assessments.
These tables are not intended to be templates, but only to illustrate the type of information that should be considered by an applicant in designing the tables for biopharmaceutic studies. Applicants should also decide whether information and results from these studies are best presented in tables, text or figures in order to aid clarity. If, for example, results are best presented in text and figures, tables might be used simply to list the studies.
2.7.2 臨床薬理の概要
2.7.2.1 背景及び概観
本項では、臨床薬理試験についての総括的な概要を示すこと。これらの試験には、ヒトでの薬物動態(PK)と薬力学(PD)を評価するための臨床試験、PKに関連するヒト細胞、組織、又は関連試料(以後、ヒト生体試料という)を用いて実施したin vitro試験が含まれる。ワクチン製品については、本項で投与量、投与スケジュール、最終製剤の選択を裏付ける免疫反応データを示すこと。適切な場合には、第2.7.1項、第2.7.3項、第2.7.4項で要約する関連データも参照して、薬物動態、薬力学、PK/PD、ヒト生体試料に関する情報の収集に用いた方法と根拠の包括的な概要を示してもよい。本項には個々の試験に関する詳細な情報を含めないこと。
本項は、まず、PK又はPDデータの解釈を助けるために実施されたヒト生体試料試験の簡潔な概観から始めること。透過性(例:腸での吸収、血液脳関門通過)、タンパク結合、肝代謝、代謝に基づく薬物相互作用に関する試験は特に重要である。次に、健康被験者及び患者におけるPK/PD関係、PKとPK/PD関係に対する内因性及び外因性要因*1の重要な影響についての試験等、医薬品のPK及びPDの特性を明らかにするために実施された臨床試験の簡潔な概観を記載すること。試験デザイン及びデータ解析の重要な側面を記載すること(例:単回又は反復投与の選択、試験対象集団、検討した内因性及び外因性要因の選択、PDエンドポイントの選択、データを収集・解析してPK又はPDを評価するために標準的方法を用いたのか、あるいはポピュレーション法を用いたのか)。
*1 外国臨床データを受入れる際に考慮すべき民族的要因についてのICH E5ガイドラインでは、被験者集団間で異なる医薬品の反応を引き起こす可能性のある要因を内因性民族的要因と外因性民族的要因に分類している。CTDにおいては、これらの分類をそれぞれ内因性要因と外因性要因と呼ぶ。
2.7.2 Summary of Clinical Pharmacology Studies
2.7.2.1 Background and Overview
This section should provide the reviewer with an overall view of the clinical pharmacology studies. These studies include clinical studies performed to evaluate human pharmacokinetics (PK), and pharmacodynamics (PD), and in vitro studies performed with human cells, tissues, or related materials (hereinafter referred to as human biomaterials) that are pertinent to PK processes. For vaccine products, this section should provide the reviewer with immune response data that support the selection of dose, dosage schedule, and formulation of the final product. Where appropriate, relevant data that are summarised in sections 2.7.1, 2.7.3 and 2.7.4 can also be referenced to provide a comprehensive view of the approach and rationale for the development of the pharmacokinetic, pharmacodynamic, PK/PD and human biomaterial database. This section should not include detailed information about individual studies.
This section should begin with a brief overview of the human biomaterial studies that were conducted and that were intended to help in the interpretation of PK or PD data. Studies of permeability (e.g., intestinal absorption, blood brain barrier passage), protein binding, hepatic metabolism, and metabolic-based drug-drug interactions are particularly relevant. This should be followed by a brief overview of the clinical studies that were carried out to characterise PK and PD of the medicinal product, including studies of PK/PD relationships in healthy subjects and patients, and relevant effects of intrinsic and extrinsic factors on PK and PK/PD relationships*. Critical aspects of study design and data analysis should be noted, e.g., the choice of the single or multiple doses used, the study population, choice of the intrinsic or extrinsic factors that were studied, the choice of PD endpoints, and whether a traditional approach or a population approach was used to collect and analyse data to assess PK or PD.
* In the ICH E5 guideline on Ethnic Factors in the Acceptance of Foreign Data, factors that may result in different responses to a drug in different populations are categorized as intrinsic ethnic factors or extrinsic ethnic factors. In this document, these categories are referred to as intrinsic factors and extrinsic factors, respectively.
2.7.2.2 個々の試験結果の要約
全ての臨床薬理試験を、通常、一覧表で提示し(第2.7.2項の付録を参照)、PK、PD及びPK/PD関係に関するin vitro又はin vivoのデータと情報が得られた重要な個々の試験の内容と結果について、簡単な説明を付け加えること。説明は、論文抄録のように簡潔なものとし、デザイン上の特徴及び重要な結果のみを記載すること。個々の試験結果及び試験間の重要な違いに着目して、試験デザインが似ているものはまとめて表記してもよい。また、本項から各総括報告書へ参照できるように配慮しておくか、電子的リンクを貼りつけておくこと。
薬力学的エンドポイントを用いた用量-反応又は濃度-反応試験の要約を本項に含めること。しかし、これらが適切に管理された試験であり、有効性又は安全性に関する重要なデータが得られる場合には、第2.7.3項又は第2.7.4項で要約し、ここでは引用するのみとする。
2.7.2.2 Summary of Results of Individual Studies
A tabular listing of all clinical pharmacology studies should generally be provided (see 2.7.2.5 Appendix), together with a narrative description of the relevant features and outcomes of each of the critical individual studies that provided in vitro or in vivo data and information relevant to PK, PD and PK/PD relationships. The narrative descriptions should be brief, e.g., similar to an abstract for a journal article, and should describe critical design features and critical results. Similar studies may be described together, noting the individual study results and any important differences among the studies. References or electronic links to the full report of each study should be included in the narratives.
Summaries of dose-response or concentration response (PK/PD) studies with pharmacodynamic endpoints should generally be included in this section. In some cases, however, when well-controlled dose-response PD or PK/PD studies provide important evidence of efficacy or safety, they should be placed in 2.7.3 or 2.7.4 as appropriate and referenced, but not summarised, here.
2.7.2.3 全試験を通しての結果の比較と解析
本項では、当該医薬品のPK、PD、PK/PD関係を特徴づけるために実施したin vitroヒト生体試料試験、PK、PD、PK/PD試験の全ての結果を用いること。これらの結果のうち、個体内及び個体間変動に関する成績、これらのPK関係に影響を及ぼす内因性及び外因性要因について示すこと。
本項では、通常は文章と表を用いて、以下の事柄に関する全ての試験データを事実に基づいて示すこと。
- In vitro薬物代謝試験及びin vitro薬物相互作用試験の結果及びその臨床的な意味。
- 標準的なパラメータの最良推定値及び変動要因の記載を含むヒトPK試験。対象疾患の患者集団及び特別な患者集団(例:小児、高齢者、腎障害や肝障害を有する患者)における用量及び用量調整を裏付けるデータに重点を置くこと。
- 単回投与時と反復投与時とのPKの比較。
- 外因性要因又は内因性要因によると思われるPK又はPDの個体間変動を検討するために、複数の試験において実施された少数サンプリングに基づく結果等のポピュレーションPK解析。
- 用量-反応又は濃度-反応関係。ここでは、重要な臨床試験で用いた投与量と投与間隔の選択を裏付けるデータに重点を置くこと。なお、添付文書案中の用法・用量を裏付ける情報については、第2.7.3.4項に示すこと。
- ヒト生体試料試験、PK試験又はPD試験から得られた結果における大きな矛盾点。
- 外国臨床データが新地域に外挿できるかどうかを決定するために実施されたPK試験(ICH E5参照)。地域又は人種間のPKデータの類似性に関する試験結果及び解析結果を本項に要約すること。PDバイオマーカーを用いる試験(しかし臨床的有効性は評価しない)は同様に本項で要約してよい。また、独立した下位の項を作り、これらの種類のデータを要約してもよい。
2.7.2.3 Comparison and Analyses of Results Across Studies
This section should use the results of all in vitro human biomaterial studies and PK, PD and PK/PD studies to characterise the PK, PD and PK/PD relationships of the drug. Results related to the inter- and intra-individual variability in these data and the intrinsic and extrinsic factors affecting these pharmacokinetic relationships should be discussed.
This section (typically with the use of text and tables) should provide a factual presentation of all data across studies pertinent to the following:
- in vitro drug metabolism and in vitro drug-drug interaction studies and their clinical implications.
- human PK studies, including the best estimates of standard parameters and sources of variability. The focus should be on evidence supporting dose and dose individualisation in the target patient population and in special populations, e.g., paediatric or geriatric patients, or patients with renal or hepatic impairment.
- comparison between single and repeated-dose PK
- population PK analyses, such as results based on sparse sampling across studies that address inter-individual variations in the PK or PD of the active drug substances that may be due to extrinsic or intrinsic factors.
- dose-response or concentration-response relationships. This discussion should highlight evidence to support the selection of dosages and dose intervals studied in the important clinical trials. In addition, information that supports the dosage instructions in the proposed labelling should be discussed in Section 2.7.3.4.
- major inconsistencies in the human biomaterial, PK, or PD database.
- PK studies that were performed to determine whether foreign clinical data could be extrapolated to the new region (see ICH E5). The result of the studies and analysis of the similarity of the PK data between regions or races should be summarised in this section. Such studies that use PD biomarkers (but do not evaluate clinical efficacy) may similarly be summarised here. An independent subsection can be created to summarise these kinds of data.
2.7.2.4 特別な試験
本項では、特定の種類の医薬品に関連する特別な種類のデータを得るために実施した試験を示すこと。免疫原性試験及び他の試験で、データがPK、PD、安全性、有効性データと関係する可能性のあるものは、その関係に係る説明を本項で要約すること。PK、PD、安全性、有効性に対して認められた影響又は考えられる影響については、本項と相互参照しながら臨床概要の適切な項に示すこと。特に懸念される安全性上の問題が認められた試験については、本項に記述する代わりに第2.7.4項「臨床的安全性の概要」に示すこと。
なお、本項の報告書は第5部第5.3.5.4項「その他の臨床試験報告書」に添付することができる。
例1:免疫原性
特異的な免疫反応が認められたタンパク製剤やその他の製剤について、免疫原性に関するデータを本項で要約すること。特定の免疫反応を誘発するためのワクチンや他の製品については、免疫原性データを有効性の項に記載すること。用いた分析法を簡単に説明し、その性能(例:感度、特異性、信頼性、確実性)に関する情報を要約すること。申請資料中の詳細な情報の記載箇所を相互参照すること。
用いた抗体分析法(例:ELISA法によるIgG、中和法)の種類別に、抗体発現率、抗体価、抗体発現時期、持続期間に関するデータを要約すること。基礎疾患、併用薬、投与量、投与期間、投与方法、製剤と抗体産生との関係を検討し要約すること。長期的に持続して投与される医薬品については、治療の中断が抗原性に及ぼす影響に関するデータを解析し要約すること。
臨床的意味があるかもしれない免疫原性との相関を分析し、要約することは特に重要である(例:特定の種類又は特定の力価の抗体の存在と、PKの変化、PDの変化、有効性の減弱、有害事象プロフィールの変化、有害事象の発現がどの程度相関すると考えられるかを判定すること)。免疫学的に仲介される可能性のある事象(例:血清病)及び投与された医薬品に対する抗体による交叉反応性内因性物質の結合により生じた事象には特に注意を払うこと。
例2:臨床微生物学
抗菌薬又は抗ウイルス薬については、活性スペクトルを検討するためのin vitro試験は、臨床的有効性に関する試験計画全体の中で重要な部分を占める。有効性決定の一環としての臨床分離株の感受性に関する特性検討を含む臨床的有効性試験は、第2.7.3項「臨床的有効性の概要」に示すこと。しかし、世界各地から得られた菌株のin vitro感受性のパターン等を評価する試験(すなわち臨床的な有効性試験とは別の意味合いの試験)は本項に示すこと。
2.7.2.4 Special Studies
This section should include studies that provide special types of data relevant to specific types of medicinal products. For immunogenicity studies and other studies in which data may correlate with PK, PD, safety, and/or efficacy data, explanations of such correlations should be summarised here. Any observed or potential effects on PK, PD, safety and/or efficacy should be considered in other appropriate sections of the Clinical Summary as well, with cross-referencing to this section. Human studies that address a specific safety issue should not be reported here, but instead should be reported in the Summary of Clinical Safety (section 2.7.4).
Example 1: Immunogenicity
For protein products and other products to which specific immunological reactions have been measured, data regarding immunogenicity should be summarised in this section. For vaccines or other products intended to induce specific immune reactions, immunogenicity data should be described in the efficacy section 2.7.3. Assays used should be briefly described and information about their performance (e.g., sensitivity, specificity, reliability, validity) should be summarised; the location in the application of detailed information should be crossreferenced.
Data regarding the incidence, titre, timing of onset and duration of antibody responses should be summarised for each type of antibody assay used (e.g., IgG by ELISA, neutralisation). Relationships of antibody formation to underlying disease, concomitant medication, dose, duration, regimen, and formulation should be explored and summarised. For drugs intended to be given as chronic, continuous therapy, any data on the impact of interruptions of therapy on antigenicity should be analysed and summarised.
It is particularly important to summarise analyses of potential clinically relevant correlates of immunogenicity, e.g., to determine the extent to which the presence of antibodies of a particular type or titer appears to correlate with alterations of PK, changes in PD, loss of efficacy, loss of adverse event profile, or development of adverse events. Particular attention should be paid to events that might be immunologically mediated (e.g., serum sickness) and events that might result from binding of cross-reactive endogenous substances by antibodies to the administered drug.
Example 2: Clinical microbiology
For antimicrobial or antiviral medicinal products, in vitro studies to characterise the spectrum of activity are an important part of the programme of studies relevant to clinical efficacy. Clinical efficacy studies that include characterisation of the susceptibility of the clinical isolates as a part of the efficacy determination should be included in Section 2.7.3, Summary of Clinical Efficacy. However, studies that evaluate such findings as the pattern of in vitro susceptibility of strains of bacteria from different parts of the world (not in the context of clinical efficacy study) would be included here.
2.7.2.5 付録
文書が読みやすくなるのであれば、図や表を適切に文章中に挿入すること。大きな表は、本項の最後に付録として添付することができる。
表2.7.2.1はPKについての薬物相互作用試験に関する情報や結果を報告するための表様式の例である。PK/PD試験、用量-反応試験、ヒト生体試料に対する影響に関する試験、ポピュレーションPK試験についても、同様の表を作成できる。この表はテンプレートではなく、申請者が臨床薬理試験をまとめる際に考慮すべき情報の種類を単に示したものである。これらの試験から得られた情報や結果が、表、文章、図を用いて最も分かりやすく示されているかについても判断すること。例えば、結果が文章及び図で良く示されている場合は、表は試験の一覧を示すためだけに用いられることになる。
その他の種類の臨床薬理試験において表を作成する場合には、以下の情報を含めることを考慮すること。
これらは単なる例示であり、どの情報を示す必要があるかは申請者が決定すること。
- ヒト生体試料を用いた代謝試験:用いた生体試料(例:ミクロソーム、肝細胞)、プローブ医薬品、代謝酵素、その酵素の寄与率と速度論的パラメータ(例:Vmax、Km)
- ヒト生体試料を用いたin vitroの薬物相互作用試験: 新医薬品を阻害する他の医薬品に関する試験については、阻害された代謝物生成、影響を受けた代謝酵素、用いた阻害剤の濃度範囲、IC50及びKi値、考えられる阻害機構を含めること。他の医薬品を阻害する新医薬品に関する試験については、これらの情報に加え、阻害された医薬品及び代謝物を含めること。
- ポピュレーションPK試験:検討された共変量、対象被験者の数及び種類、統計パラメータの要約、平均(±標準偏差)PKパラメータの最終推定値
2.7.2.5 Appendix
Tables and figures should be embedded in the text of the appropriate sections when that enhances the readability of the document. Lengthy tables can be provided in the appendix at the end of the Section.
Table 2.7.2.1 is provided as an example of a tabular format for reporting information and results related to pharmacokinetic drug-drug interaction studies. Similar tables could be prepared for PK/PD studies, dose-response studies, studies of effects on human biomaterials, and population PK studies. This table is not intended to be a template, but only to illustrate the type of information that should be considered by sponsors in designing their own tables. Applicants should also decide whether information and results from clinical pharmacology studies are best presented in tables, text or figures in order to aid clarity. If, for example, results are best presented in text and figures, the tables might simply list the studies.
In designing tables, if any, for various types of other clinical pharmacology studies such as those listed below, applicants should consider including the following types of information.
These examples are for illustrative purposes only and the sponsor should decide which information needs to be presented.
- metabolism studies using human biomaterials: biomaterials used (e.g., microsomes, hepatocytes), probe drugs, enzymatic pathways and % contribution and relevant kinetic parameters (e.g., Vmax, Km).
- in vitro studies of drug-drug interactions using human biomaterials: for studies of other drugs inhibiting the new drug, the metabolite(s) inhibited, enzymatic pathways affected, range of inhibitor concentrations used, IC50 and Ki values and proposed mechanism of inhibition should be included. For studies of the new drug inhibiting other drugs, the drugs and metabolites inhibited should be included, along with the information mentioned above.
- population PK studies: co-variates studied, number and type of subjects or patients studied, summary statistical parameters and final estimates of mean (± standard deviation) PK parameters.
2.7.3 臨床的有効性の概要
第2.7.3項は適応症ごとにおこすこと。ただし、適応症が密接に関連する場合はひとつの項目にまとめてもよい。本項が複数のセクションとなる場合、2.7.3 肺炎、2.7.3 尿路感染症等と表示すること。
2.7.3 Summary of Clinical Efficacy
A separate Section 2.7.3 should be provided for each indication, although closely related indications can be considered together. When more than one Section 2.7.3 is submitted, the sections should be labelled 2.7.3 pneumonia, 2.7.3 URI, etc.
2.7.3.1 背景及び概観
本項では、個々の申請適応症における有効性を評価した比較対照試験及びその他の試験の実施計画を記述すること。これらの試験における安全性評価の結果は、第2.7.4項「臨床的安全性の概要」で考察すること。
本項は、有効性を評価するために実施された比較対照試験のデザインを概観することから始めること。対象となる試験には、用量反応試験、比較対照試験、長期有効性試験、特別な患者集団における有効性試験が含まれる。無作為化手順、盲検化手順、対照治療の選択、対象患者の選択、クロスオーバー法やランダム治療中止デザイン等のデザイン特性、run-in期間の設置、その他のエンリッチメント法(強化法)、採用したエンドポイント、試験期間、事前に計画した解析方法等の重要な試験デザインの特徴について考察すること。本項は臨床的評価に焦点を置いたものであるが、適宜、非臨床データや臨床薬理学データも参照し、有効性に関連する臨床使用経験を包括的に要約してもよい。ただし、本項には個々の試験の詳細を記述しないこと。
2.7.3.1 Background and Overview of Clinical Efficacy
This section should describe the program of controlled studies and other pertinent studies in the application that evaluated efficacy specific to the indication(s) sought. Any results of these studies that are pertinent to evaluation of safety should be discussed in Section 2.7.4, Summary of Clinical Safety.
The section should begin with a brief overview of the design of the controlled studies that were conducted to evaluate efficacy. These studies include dose-response, comparative efficacy, long-term efficacy, and efficacy studies in population subsets. Critical features of study design should be discussed, e.g., randomisation, blinding, choices of control treatment, choice of patient population, unusual design features such as crossover or randomised withdrawal designs, use of run-in periods, other methods of “enrichment”, study endpoints, study duration, and prespecified plans for analysis of the study results. Although this section is intended to focus on clinical investigations, nonclinical data and clinical pharmacology data may also be referenced as appropriate to provide a comprehensive summary of human experience related to efficacy. This section should not include detailed information about individual studies.
2.7.3.2 個々の試験結果の要約
申請医薬品の有効性を検討した(又は検討を目的とした)全ての試験を、通常、一覧表で提示し(第2.7.3項の付録を参照)、重要な試験については説明的な記述を加えること。説明は、論文抄録のように簡潔なものとし、デザイン上の特徴及び重要な結果のみを記載すること。試験デザインが似ているものはまとめて記載してもよいが、その場合、結果がどの試験のものかが分かるように記載し、試験間の結果に重要な差異があれば示すこと。また、安全性評価上重要な試験については、どの程度被験者が被験薬又は対照薬に曝露されたか、安全性データをどのように収集したかについて説明すること。これらの記述は、総括報告書の概要(シノプシス)(ICH E3)から抜粋してもよい。また、本項から各総括報告書へ参照できるように配慮しておくか、電子的リンクを貼りつけておくこと。
ある特定の外国臨床データが新地域に外挿できるかどうかを評価するために実施した試験等、臨床的エンドポイントを用いたブリッジング試験(ICH E5を参照)に関する記述は、本項に含めること。必要であれば、そのブリッジング試験の結果について、外国試験での有効性と安全性を外挿するうえで有用と思われるその他の情報(例:PK及びPDデータ)と共に検討すること。検討により得られた結論は第2.7.3.3.2項「全有効性試験の結果の比較検討」の最初に示し、検討結果の全文は第5部に添付すること。
2.7.3.2 Summary of Results of Individual Studies
A tabular listing of all studies that provided (or were designed to provide) information relevant to product efficacy should generally be provided (see the section 2.7.3.6 Appendix), together with narrative descriptions for important studies. The narrative descriptions should be brief, e.g., similar to an abstract for a journal article, and should describe critical design features and critical results. Similar studies may be described together, noting the individual study results and any important differences among the studies. For studies that also contributed significantly to the safety analysis, study narratives should include information about the extent of exposure of study subjects to the test drug or control agent, and how safety data were collected. These narratives can be abstracted from the synopses of the clinical study reports (ICH E3). References or electronic links to the full report of each study should be included in the narratives.
Narratives of any bridging studies using clinical endpoints, i.e., certain studies intended to evaluate the ability to extrapolate certain types of foreign clinical data to the new region (see ICH E5), should be included in this section. An analysis of the results of such studies, together with other information (e.g., PK and PD data) that addresses the ability to extrapolate the efficacy and safety results of foreign studies, should be performed if necessary. The conclusions of such an analysis should be noted at the start of Section 2.7.3.3.2, Comparison of Efficacy Results of All Studies, and the full report of the analysis should be provided in Module 5.
2.7.3.3 全試験を通しての結果の比較と解析
第2.7.3.3項中のサブセクションでは、文章、図、表を適切に用い(第2.7.3項付録を参照)、当該医薬品の有効性を特徴づける全てのデータを要約すること。この要約には、最終的な結論を裏付けているかどうかにかかわりなく、全てのデータの解析結果が含まれていなければならず、従って、関連する試験が互いにどの程度結論を確立するうえで有用かどうかも考察すること。有効性に関するデータ間に重大な不整合があれば記載し、追加検討を要する事柄を明らかにすること。
本項における有効性に関する検討方法は通常二種類あり、一つは個々の試験結果の比較であり、もう一つは複数試験からのデータを併せた分析である。本項に記載できない詳細な検討結果は別途第5部第5.3.5.3項に添付すること。
さらに、添付文書中の用法・用量の内容を裏付ける第2.7.2項に含まれるデータ等の重要な科学的根拠を示すこと。対象となるデータとしては、推奨する用量及び投与間隔、個々の患者における用量調整、特別な患者集団における用量変更の必要性(小児、高齢者、肝障害、又は腎障害のある患者等)、用量-反応又は濃度-反応関係(PK/PD)等が含まれる。
2.7.3.3 Comparison and Analyses of Results Across Studies
Using text, figures, and tables as appropriate (see the section 2.7.3.6 Appendix), the subsections of 2.7.3.3 should summarise all available data that characterise the efficacy of the drug. This summary should include analyses of all data, irrespective of their support for the overall conclusion and should, therefore, discuss the extent to which the results of the relevant studies do or do not reinforce each other. Any major inconsistencies in the data regarding efficacy should be addressed and any areas needing further exploration should be identified.
The section will generally utilise two kinds of analyses: comparison of results of individual studies, and analysis of data combined from various studies. Details of analyses that are too extensive to be reported in a summary document should be presented in a separate report, to be placed in Module 5, Section 5.3.5.3.
This section should also cross-reference important evidence from section 2.7.2, such as data that support the dosage and administration section of the labelling. These data include dosage and dose interval recommended, evidence pertinent to individualisation of dosage and need for modifications of dosage for specific subgroups (e.g., paediatric or geriatric subjects, or subjects with hepatic or renal impairment), and data relevant to dose-response or concentration response (PK/PD) relationships.
2.7.3.3.1 試験対象集団
有効性を検討した全ての試験における被験者の人口統計学的及び他の基準値の特性を記載する。含むべき内容は以下のとおり。
- 被験者における疾患の特性(例:重症度、罹病期間)と前治療、選択/除外基準。
- 試験間又は試験グループ間における試験対象集団の基準値特性の差異。
- 主たる有効性の評価対象となった試験対象集団と市販後に使用が予想される患者集団との差異。
- 試験から脱落した患者の数、中止時期(治験期間中あるいは追跡期間中の試験日・来院時期)、投与中止理由についての評価。
表形式により試験対象集団を併記して比較することは有用と思われる。
2.7.3.3.1 Study Populations
The demographic and other baseline characteristics of patients across all efficacy studies should be described. The following should be included:
- the characteristics of the disease (e.g., severity, duration) and prior treatment in the study subjects, and study inclusion/exclusion criteria
- differences in baseline characteristics of the study populations in different studies or groups of studies.
- any differences between populations included in critical efficacy analyses and the overall patient population that would be expected to receive the drug when it is marketed should be noted.
- assessment of the number of patients who dropped out of the studies, time of withdrawal (a defined study day or visit during treatment or follow up period), and reasons for discontinuation.
Tabular presentations that combine and compare study populations across studies may be useful.
2.7.3.3.2 全有効性試験の結果の比較検討
ある特定の外国臨床データを新地域に外挿できるかどうかを検討するために実施した試験等、臨床的エンドポイントを用いたブリッジング試験(ICH E5を参照)に関する要約は、本項に記載すること。地域間における有効性データの類似性について検討した結果、又は新地域における有効性データの外挿を可能にする情報も本項にて要約する。これらの要約のため本項にサブセクションをおこしてもよい。
結論が明確でない試験や否定的な試験も含め、有効性の評価を目的とした全ての試験の結果を要約し比較すること。エンドポイント、対照群、試験期間、統計解析方法、被験者集団、投与量等で重要な試験デザイン上の差異があれば明示すること。
試験間の比較は、プライマリーエンドポイントとして事前に規定した項目に焦点を合わせて行うこと。しかし、プライマリーエンドポイントが試験間で共通でない場合や測定点が同一でない場合は、全ての試験に共通して測定された重要データを用いて比較することが有用であろう。経時的な結果が重要だと思われるのであれば、各試験における経時的な変化を図示してもよい。
治療効果に関する信頼区間を表示し、点推定値を解釈するための助けとすること。ベースラインからの変化量においてプラセボと治験薬との間に差が認められる場合、プラセボ群又は実薬対照群(用いた場合)を含む各々の治療群でのベースライン値と治療効果の大きさを、基本的には表形式で、あるいは文章に図を添付した形式で示すこと。実薬対照を用いた試験の目的が、同等性又は非劣性の証明にある場合、群間差の値や比率を信頼区間と共に示すこと。結果は、事前に定義した同等性又は非劣性の基準により評価し、その基準の理論的根拠と試験の分析感度を検討した根拠を示すこと(ICH E10を参照)
試験デザインが似ているにもかかわらず試験結果に重大な相違が認められた場合は、その違いを詳述し考察すること。また、相違に関係したと思われる要因を試験間で比較し示すこと。
メタアナリシスが実施されている場合、その解析がプロトコルに基づきなされたものか、試験後に実施されたものかを明確にすること。試験デザインの比較や試験対象集団の違い、あるいは有効性評価法の違いを記述して、結果や結論の類似性及び妥当性の評価ができるようにすること(ICH E9を参照)。メタアナリシスの方法や結果に関する詳細な説明は、通常、別の報告書として提出すること(第5部第5.3.5.3項)。
2.7.3.3.2 Comparison of Efficacy Results of all Studies
The results of any bridging studies using clinical endpoints, i.e., certain studies used to evaluate the ability to extrapolate certain types of foreign clinical data to the new region (see ICH E5), should be summarised in this section. An analysis of the similarity of efficacy in subjects between regions, as well as any other information that may support extrapolation of the efficacy data to the new region, should be summarised here. An independent subsection can be created to summarize these kinds of data.
The results from all studies designed to evaluate the drug’s efficacy should be summarised and compared, including studies with inconclusive or negative results. Important differences in study design such as endpoints, control group, study duration, statistical methods, patient population, and dose should be identified.
Comparisons of results across studies should focus on pre-specified primary endpoints. However, when the primary endpoints involved different variables or time points in different efficacy studies, it may be useful to provide cross-study comparisons of important data elements that were obtained in all studies. If results over time are important, results of studies may be displayed in a figure that illustrates the change over time in each study.
Confidence intervals for treatment effects should be given to aid in the interpretation of point estimates. If differences are shown between placebo and test drugs in the change from baseline, the baseline values and the magnitude of effect in all treatment groups, including placebo and active controls (if used), should generally be presented in the table or in text accompanying a figure. If the objective of an active control trial was to show equivalence or non-inferiority, the difference or the ratio of outcomes between treatments should be given with the confidence interval. The results should be evaluated by using the predefined criteria for defining equivalence or non-inferiority and the rationale for the criteria and support for the determination that the study (studies) had assay sensitivity should be provided (see ICH E10).
Important differences in outcomes between studies with a similar design should be delineated and discussed. Cross-study comparisons of factors that may have contributed to differences in outcomes should be described.
If a meta-analysis of the clinical studies is performed, it should be clear whether this analysis is conducted according to a predefined protocol or is a post hoc exercise. Any differences in trial designs or populations, or in efficacy measurements between trials should be described to allow assessment of the relevance and validity of the results and conclusions (See ICH E9). A detailed description of the methodology and results of the meta-analysis should generally be submitted in a separate report (section 5.3.5.3 of Module 5).
2.7.3.3.3 部分集団における結果の比較
本項では、特定の患者集団における有効性に関する個々の試験の結果又は全試験を通じての概括的な評価を要約すること。部分集団にて比較する目的は、対象となる全ての部分集団、特に、懸念される特別な理由がある部分集団で、主張する治療効果が一貫して得られているかどうかを示すことにある。比較をすることで、検討や考察をさらに必要とする有効性のバラツキを明らかにすることにもなるだろう。
ただし、こうした分析には限界もあることを認識しておくこと(ICH E9)。また、かかる比較は、特定の効能・効果の根拠そのものを生み出すためのものではなく、全体の結果が好ましくない状況下で有効性の根拠を補強するためのものでもないことに注意すること。
個々の試験における症例数が十分でない場合、複数の試験をまとめて分析を行い、有効性に影響する主要な人口統計学的特性(年齢、性別、人種)、事前に定めた又は関連する内因性/外因性要因(例:重症度、前治療、合併症、併用薬、飲酒、喫煙、体重)について評価すること。さらに、一般的に懸念される要因(例:高齢者)や当該医薬品の薬理作用から考えられる要因、又は開発の初期段階にて生じた特別な要因等も評価すること。申請適応症から小児も対象となり得る場合、小児における有効性を分析し記述すること。詳細な解析が実施された場合は、その報告書を第5部に添付し、要約をこの項に記載してよい。
2.7.3.3.3 Comparison of Results in Sub-populations
The results of individual studies or overview analyses of efficacy in specific populations should be summarised in this section. The purpose of these comparisons should be to show whether the claimed treatment effects are observed consistently across all relevant subpopulations, especially those where there are special reasons for concern. The comparisons may highlight apparent variations in efficacy that require further investigation and discussion.
The limitations of such analyses, however, should be recognised (ICH E9), and it is important to note that their purpose is not to provide the basis for specific claims, nor to attempt to improve the evidence of efficacy in situations where the overall results are disappointing.
Given the limited sample sizes in individual studies, analyses across multiple studies should be performed to evaluate effects of major demographic factors (age, sex, and race) and of other predefined or relevant intrinsic and extrinsic factors (e.g., disease severity, prior treatment, concomitant illness, concomitant drugs, alcohol, tobacco, and body weight) on efficacy. Factors of special interest may arise from general concerns (e.g., the elderly) or from specific issues that are related to the pharmacology of the drug or that have arisen during earlier drug development. Efficacy in the paediatric population should be routinely analysed in applications for a proposed indication that occurs in children. Depending on the data set, if extensive, detailed efficacy analyses are performed, they can be placed in Module 5, with the results of those analyses reported here.
2.7.3.4 推奨用法・用量に関する臨床情報の解析
本項では、有効性における用量-反応又は血中濃度-反応関係(用量-血中濃度関係を含む)、そして投与量の選択及び投与間隔の選択のために使われた全ての資料の概要と分析結果を記載すること。非臨床試験からの関連するデータを引用し、薬物動態試験、臨床薬理試験、比較対照試験、非対照試験を要約して、用量-反応又は血中濃度-反応関係を説明すること。第2.7.2.2項で要約した薬物動態試験と薬力学試験については、その要約を繰り返し記述せず、参照しながら内容を引用する方が適切な場合もある。
これらのデータがいかに推奨する用法・用量を裏付けているかについての解釈は「臨床に関する概括評価」に記載されるものであるが、本項では、当該用法・用量(開始用量と最大用量、用量漸増法、用量個別化のための注意事項を含む)を推奨するために使用された個々の試験の結果と試験を総括的に分析し得られた結果を要約すること。また、非線形性の薬物動態、効果発現の遅延、耐薬性の発現、酵素誘導等による比較的単純な用量-反応や血中濃度-反応関係からの逸脱についてもこの項に記載すること。
患者の年齢、性別、人種、疾患、その他の要因により生じる用量-反応関係の差異を示すデータを記載すること。薬物動態的、薬力学的反応の差異を示すデータも本項にて提示するか、第2.7.2項を引用すること。差異が見られない場合でも、どのような検討を行ったかその方法を記載すること(例:部分集団における特別な試験、部分集団別有効性分析、被験薬の血中濃度測定)。
2.7.3.4 Analysis of Clinical Information Relevant to Dosing Recommendations
This section should provide an integrated summary and analysis of all data that pertain to the dose-response or blood level-response relationships of effectiveness (including dose-blood level relationships), and thus have contributed to dose selection and choice of dose interval. Relevant data from nonclinical studies may be referenced, and relevant data from pharmacokinetic studies, other clinical pharmacology studies, and controlled and uncontrolled clinical studies should be summarised to illustrate these dose-response or blood level-response relationships. For pharmacokinetic and pharmacodynamic studies from which data have been summarised in Section 2.7.2.2, it may be appropriate to draw upon those data in this summary while cross-referencing the summaries in Section 2.7.2.2, without repeating those summaries.
While the interpretation of how these data support specific dosing recommendations should be supplied in the Clinical Overview document, the individual study results and any cross-study analyses that will be used to support the dosing recommendations (including the recommended starting and maximal doses, the method of dose titration, and any other instructions regarding individualisation of dosage) should be summarised here. Any identified deviations from relatively simple dose-response or blood-level response relationships due to non-linearity of pharmacokinetics, delayed effects, tolerance, enzyme induction, etc. should be described.
Any evidence of differences in dose-response relationships that result from a patient’s age, sex, race, disease, or other factors should be described. Any evidence of different pharmacokinetic or pharmacodynamic responses should also be discussed, or discussions in Section 2.7.2 can be cross-referenced. The ways in which such differences were looked for, even if no differences were found, should be described (e.g., specific studies in subpopulations, analysis of efficacy results by subgroup, or blood level determinations of the test drug).
2.7.3.5 効果の持続、耐薬性
効果の持続に関する情報があれば要約すること。長期投与時のデータが得られている被験者数と曝露期間を示すこと。耐薬性(経時的な効果の減弱)のデータを示すこと。経時的な投与量の変更と長期投与時の有効性との間に明らかな関係が存在していないか検討することは有用であろう。
本項での主眼は、長期有効性のデータを収集するためにデザインされた比較対照試験に置き、比較対照試験終了後の継続投与オープン試験等の厳密でない試験とは明確に区別すること。このような区別は、耐薬性や効果に対する投与終了の影響等を検討する特別な試験でも行うこと。安全性に関連した離脱作用や反跳作用についてのデータは、安全性の項に提示すること(第2.7.4項を参照)。
長期有効性試験に関して、治療中止や他療法への切り替えが有効性の評価にどのような影響を与えたかを考察すること。これらの事柄は短期の試験についても重要であるので、適切と考えられる場合には、同様に考察すること。
2.7.3.5 Persistence of Efficacy and/or Tolerance Effects
Available information on persistence of efficacy over time should be summarised. The number of patients for whom long-term efficacy data are available, and the length of exposure, should be provided. Any evidence of tolerance (loss of therapeutic effects over time) should be noted. Examination of any apparent relationships between dose changes over time and long-term efficacy may be useful.
The primary focus should be on controlled studies specifically designed to collect long-term efficacy data, and such studies should be clearly differentiated from other, less rigorous, studies such as open extension studies. This distinction also applies to specific studies designed for evaluation of tolerance and withdrawal effects. Data concerning withdrawal or rebound effects pertinent to product safety should be presented in the safety section (see section 2.7.4).
In long-term efficacy trials, the effect of premature discontinuation of therapy or switching to other therapies upon the assessment of the results should be considered. These issues might also be important for short term trials and should be addressed when discussing the results of these trials, if appropriate.
2.7.3.6 付録
文書が読みやすくなるのであれば、図や表を適切に文章中に挿入すること。大きな表は、本項の最後に付録として添付することができる。
表には、有効性の評価に関連した全ての試験(中止した試験、進行中の試験、理由にかかわらず有効性を示せなかった試験、公表文献のみ利用可能な試験、総括報告書(ICH-E3)にまとめられた試験、「簡略化された報告書」にまとめられた試験を含む)を示し、それぞれの試験における最も重要な結果を記載すること。ただし、進行中の試験における計画外の中間解析は、原則として必要でないし、薦められない。複数の適応症による申請において第2.7.3項が複数存在する場合、通常、各項ごとに表を付録として添付すること。
降圧薬の表を例示するが、例示が全ての申請において当てはまるわけではない。一般的に、申請にあたっては、その薬効分類又は実施された試験用に作成された表や図が必要となる。
表2.7.3.1 臨床的有効性及び安全性試験の要約
表2.7.3.2 有効性試験の結果
2.7.3.6 Appendix
Tables and figures should be embedded in the text of the appropriate sections when that enhances the readability of the document. Lengthy tables can be provided in the appendix at the end of the Section.
Tables should identify all studies pertinent to the evaluation of efficacy (including studies that were terminated or are not yet completed, studies that failed to show effectiveness for any reason, studies available only as publications, studies reported in full technical reports (ICH E3), and studies described in abbreviated reports); and should provide the most important results of those studies. Note, however, that unplanned interim analyses on ongoing studies are generally not needed or encouraged. When more than one section 2.7.3 is provided for an application with more than one indication, usually each section should have its own appendix with tables.
Illustrative tables for an antihypertensive drug are provided, but these examples will not be relevant to every application. In general, applications will require tables and/or figures that are developed specifically for the particular drug class and the studies that were carried out.
Table 2.7.3.1 Description of Clinical Efficacy and Safety Studies
Table 2.7.3.2 Results of Efficacy Studies
2.7.4 臨床的安全性の概要
本項では、個々の総括報告書及び他の関連する報告書(例:一部地域にて通常提出されている安全性に関する統合解析)の結果をまとめ、対象となる患者集団における申請医薬品の安全性に関するデータを要約すること。安全性関連データの示し方は、次の三つのレベルで考察することができる(ICH E3)。
- 曝露状況(投与量、投与期間、患者数、患者のタイプ)を検討し、データベースからどの程度の安全性評価が可能なのかを決定すること。
- 比較的よくみられる有害事象や臨床検査値の変化を明確にし、妥当な方法で分類し、それらの発現に関する要約を行うこと。
- 重篤な有害事象(ICH E2Aによる定義)及びその他の重要な有害事象(ICH E3による定義)を明確にし、それらの発現について要約すること。これら事象の頻度を、特に長期にわたり使用される可能性がある医薬品については、経時的に検討すること。
全ての臨床的安全性データを解析し、得られた申請医薬品の安全性プロフィールは、図表を使って詳細に、明確に、客観的に概説すること。
2.7.4 Summary of Clinical Safety
This section should be a summary of data relevant to safety in the intended patient population, integrating the results of individual clinical study reports as well as other relevant reports, e.g., the integrated analyses of safety that are routinely submitted in some regions.
The display of safety-related data can be considered at three levels (ICH E3):
- The extent of exposure (dose, duration, number of patients, type of patients) should be examined to determine the degree to which safety can be assessed from the database.
- The more common adverse events and changes in laboratory tests should be identified and classified, and their occurrence should be summarised.
- Serious adverse events (defined in ICH E2A) and other significant adverse events (defined in ICH E3) should be identified and their occurrence should be summarised. These events should be examined for frequency over time, particularly for drugs that may be used chronically.
The safety profile of the drug, described on the basis of analysis of all clinical safety data, should be outlined in a detailed, clear, and objective manner, with use of tables and figures.
2.7.4.1 医薬品への曝露
2.7.4.1.1 総括的安全性評価計画及び安全性試験の記述
本項では、安全性に関する評価計画の全体を概括すること。その際、非臨床データ、同じ薬効分類の薬剤に共通する作用、そして臨床では、安全性データの元となった資料(比較対照試験、オープン試験等)について特に配慮したり観察した事柄に言及すること。記載は、一般的には、一覧表形式にて安全性データの元となった全ての臨床試験を適切にグループ化し、まとめること(第2.7.4項付録を参照)。有効性と安全性を共に評価した試験や安全性情報を得た非対照試験のみでなく、安全性上の特別な問題点を検討した試験も列記すること。例として、二つの異なる治療法を用いて特定の有害事象の発現率を比較検討した試験や、特別な人口統計学的特性について検討した安全性試験、離脱症状や反跳現象を検討した試験、その他、特定の有害事象(例:鎮静、性的機能、運転に対する影響、同一薬効群の薬剤で一般的に認められる有害事象がないこと)を評価した試験等が挙げられる。当該申請にて承認を求めていない適応症についての試験や進行中の試験も、安全性の解析に有用であるならば、本項の表に含めること。
本項では、これらの試験について文章で説明すること。ただし、有効性と安全性データの両方を含む試験の説明は第2.7.3.2項に記載し、本項ではそれを参照すること。説明の詳しさは、審査担当者が被験薬又は対照薬への曝露状況を理解でき、またどのように安全性データを収集したかが理解できるようなものであること(例:収集方法及び個々の試験における被験者観察の範囲)。試験一つ一つでなく、グループ化して解析した場合、その旨を記載し、一つの説明にまとめてもよい。
2.7.4.1 Exposure to the Drug
2.7.4.1.1 Overall Safety Evaluation Plan and Narratives of Safety Studies
The overall safety evaluation plan should be described briefly, including special considerations and observations concerning the nonclinical data, any relevant pharmacological class effects, and the sources of the safety data (controlled trials, open studies, etc). A tabular listing of all clinical studies that provided safety data, grouped appropriately, should generally be provided (see the section 2.7.4.7 Appendix). In addition to studies that evaluated efficacy and safety, and uncontrolled studies that generate safety information, this section includes studies that consider special safety issues. Examples would include studies to compare particular adverse event rates for two therapies, to assess safety in particular demographic subsets, to evaluate withdrawal or rebound phenomena, or to evaluate particular adverse events (e.g., sedation, sexual function, effects on driving, absence of a class adverse effect). Studies in indications for which approval is not being sought in the current application and ongoing studies would also be included here if they contribute to the safety analysis.
Narrative descriptions of these studies should be provided here, except that narrative descriptions for studies that contributed both efficacy and safety data should be included in Section 2.7.3.2 and cross-referenced here. The narratives should provide enough detail to allow the reviewer to understand the exposure of study subjects to the test drug or control agent, and how safety data were collected (including the methods used and the extent of safety monitoring of the subjects enrolled in the individual studies). If some studies are not analysed separately but are grouped for safety analysis, that should be noted, and a single narrative description can be provided.
2.7.4.1.2 全般的な曝露状況
全ての臨床開発の相における治験薬への曝露状況を表形式(第2.7.4項付録中の例を参照)にまとめ、適切な説明をつけて概括すること。表には、異なるタイプの試験、異なる投与量、投与経路、投与期間別に曝露された被験者数を示すこと。もしも、投与量、曝露期間の種類が多い場合、当該医薬品に適切と思われる方法でグループ化してよい。例えば、各投与量、投与量の範囲、投与期間ごとに、1日以下、2日から1週、1週から1ヵ月、1ヵ月から6ヵ月、6ヵ月から1年、1年超(ICH E3)といった曝露期間別に被験者数を表示することができる。申請適用におけるその医薬品の安全性評価に特別な意味を持つと思われる診断別部分集団や特定の併用療法を受けた患者群を明確にすることが重要となる申請もある。
この表に記載される投与量は、投与された最大投与量、最も長期間投与された用量、平均1日用量等が考えられる。累積投与量が適切である場合もある。表示法としては、必要に応じ、実際に投与された1日用量又はmg/kgあるいはmg/m2単位とする。有害事象や臨床検査値の変化との相関性を検討するため薬物濃度(例:有害事象発現時の濃度、最大血漿中濃度、AUC)も、もしデータがあれば、有用である。
安全性解析には、治験に組み入れられ、少なくとも1回治験薬の投与を受けた全ての被験者を含めるものと想定している。そうでない場合には、その旨説明すること。
2.7.4.1.2 Overall Extent of Exposure
A table (see example provided in the section 2.7.4.7 Appendix) and appropriate text should be generated to summarise the overall extent of drug exposure from all phases of the clinical study development programme. The table should indicate the numbers of subjects exposed in studies of different types and at various doses, routes, and durations. If a large number of different doses and/or durations of exposure were used, these can be grouped in a manner appropriate for the drug. Thus, for any dose or range of doses, duration of exposure can be summarised by the number of subjects exposed for specific periods of time, such as 1 day or less, 2 days to 1 week, 1 week to 1 month, 1 month to 6 months, 6 months to 1 year, more than 1 year (ICH E3). In some applications it may be important to identify diagnostic subgroups and/or groups receiving specific concomitant therapies deemed particularly relevant to safety assessment in the intended use.
The dose levels used for each subject in this presentation could be the maximum dose received by that subject, the dose with longest exposure, and/or the mean daily dose, as appropriate. In some cases, cumulative dose may be pertinent. Dosage may be given as the actual daily dose or on a mg/kg or mg/m2 basis, as appropriate. If available, drug concentration data (e.g., concentration at the time of an adverse event, maximum plasma concentration, area under curve) may be helpful in individual subjects for correlation with adverse events or changes in laboratory variables.
It is assumed that all subjects who were enrolled and received at least one dose of the treatment are included in the safety analysis; if that is not so, an explanation should be provided.
2.7.4.1.3 治験対象集団の人口統計学的特性及びその他の特性
要約表を作成し、開発期間中、治験薬に曝露された集団の人口統計学的特性(表2.7.4.2)について概観すること。年齢幅を決める時は、ICH E7 (特別な患者集団における試験:高齢者) 及びICH E11 (小児集団における医薬品の臨床開発について)での議論を考慮すること。もし、比較対照試験における人口統計学的特性別グループでの曝露が全体の曝露との比較において異なる場合、まとめの表は別にする方が便利なこともある。
さらに、一つ以上の表を用い、試験集団全体の特性及び特殊な特性を有する被験者数を示すこと。特殊な特性には次のものが含まれる。
- 疾患の重症度
- 入院
- 腎機能障害
- 合併症
- 特定の医薬品の併用
- 治験実施地域
これらの特性が比較対照試験と全体のデータベースの間で異なる分布を示す場合、一般的に両グループを別の表で示すとよい。
被験薬とプラセボ、対照薬との間で人口統計学的特性上の不均衡があれば表に説明をつけること。特に、その不均衡が安全性の評価結果に違いをもたらす可能性がある場合には、説明をつけること。
また、特定の患者(合併症、疾患の重症度、併用医薬品)を除外した場合、その事実を記載すること。
人口統計学的特性は、適応症ごとに一覧表としてまとめること。ただし、適応症が相互に密接に関連している場合、被験者特性からみたリスクが同じと考えられるならば、一緒に扱ってよい。
2.7.4.1.3 Demographic and Other Characteristics of Study Population
A summary table should provide the reader with an overview of the demographic characteristics (Table 2.7.4.2) of the population that was exposed to the therapeutic agent during its development. Choice of age ranges used should take into account considerations discussed in ICH E7 [Studies in Support of Special Populations: Geriatrics] and ICH E11 [Clinical Investigation of Medicinal Products in the Paediatric Population]. If the relative exposure of demographic groups in the controlled trials differed from overall exposure, it may be useful to provide separate tables.
In addition, one or more tables should show the relevant characteristics of the study population, and the numbers of subjects with special characteristics. Such characteristics could include:
- Severity of disease
- Hospitalisation
- Impaired renal function
- Concomitant illnesses
- Concomitant use of particular medications
- Geographical location
If these characteristics are distributed differently in controlled trials versus the overall database, it will generally be useful to present tables on both groupings.
The text accompanying the table(s) should mention any imbalance(s) between the drug and placebo and/or comparator regarding any of the above demographic characteristics, particularly if they could lead to differences in safety outcomes.
If certain subjects were excluded from studies (concomitant illness, severity of illness, concomitant medications), this fact should be noted.
Separate demographic tables should be provided for every indication, although closely related indications can be considered together, if study subject characteristics are such that risks are believed to be the same.
2.7.4.2 有害事象
2.7.4.2.1 有害事象の解析
本項には、有害事象の頻度に関するデータを文章及び表形式にて記載すること。文章によるまとめは第2.7.4.2.1項中の適切なサブセクションに示し、文中に含めない独立した表は第2.7.4項の付録に添付すること。
治療開始後に発現した又は悪化した全ての有害事象(「治療により発現した兆候及び症状」、治療前には見られなかった事象、治療前からあったが治療中に悪化した事象)について作表し、事象の種類とそれぞれでの被験者数、頻度を被験薬、実対照薬、プラセボの別に要約すること。その表に投与量を示してもよいし、変更して、重症度別や(開始後)発現時期別、因果関係別に発現頻度等を示してもよい。
もし、記載しようとする安全性データがごく少数の治験に基づく場合(例:一つか二つの試験のみ)や被験者群の特徴が非常に異なっている場合には、データを試験別にまとめるほうが適切であることが多い。データが少数試験によらない場合には、いくつかの試験を合わせ、データを併合し、推定値の精度や差に対する感度を増すよう考慮してみること。
試験を合わせ、データを併合することは有用であることが多いが、解釈が難しくなったり差異を見えにくくしたりすることもあるので、注意して取り扱うこと。差異が明確である場合には、試験ごとにデータをまとめる方が適切である。考慮すべき事柄としては次のようなものがある。
- データを併合するのが適切と考えられるのは、投与量、投与期間、有害事象の評価方法及び患者集団等の試験デザインが類似している場合である。
- ある特定の有害事象の発現率が併合する個々の試験においてかなり違う場合、併合解析から得られる推定値の意義はあまりない。
- 他の試験と比較し、有害事象のパターンが違っている試験は別途まとめること。
- 解析の程度は、見られた有害事象の重篤性と因果関係の根拠の強さによる。因果関係のある有害事象の発現率、重篤な有害事象の発現率、又は投与中止あるいは用法・用量の変更をもたらした事象の発現率に明らかな違いがあれば詳細に検討を必要とするが、その他の有害事象について詳細に解析する意味はない。
- どのような被験者が臨床検査の極端な異常値(「アウトライアー」)を経験するか検討することは、特定の有害事象に対するハイリスク集団を明らかにするのに有用であろう。
次のような試験は併合し、安全性解析を行ってよい。
- 全ての比較対照試験又は比較対照試験の部分集団。例えば、全てのプラセボ対照試験、何らかの実薬対照を用いた試験、特定の実薬対照を用いた試験、特定の適応症について検討した試験(異なる患者集団にて実施された試験)がその対象になる。これらのグループ化は、比較的よく見られる有害事象についての最も重要な情報源であると考えられ、これにより、自然発生的な事象と治験薬と関連のある事象とを区別することができる。有害事象の発現率については対照薬群と被験薬群との間で比較すること。
- 健康被験者を対象とした短期試験を除いた全ての試験。比較的稀な事象を評価するには、このグループ化が最も適している。
- 特定の投与経路、投与方法、特定の併用療法を用いた全ての試験。
- チェックリストや直接質問に基づき有害事象を調査した試験、又は自発報告に基づく試験。
- 試験を実施した地域ごとに併合した結果。
これらのうち、最初の二つのグループ化はほぼ全ての場合に有用である。しかし、他のグループ化の意義は薬剤により異なるので、個々の試験結果を精査した後に決める方がよい。いかなる試験方法であれ、試験それぞれにおける発現率は実際の使用における発現をおおよそ表現したものにすぎないことを認識すること。
いくつかの試験のデータを併合する場合は、その方法を選択した根拠を説明すること。一般的な方法としては、併合した試験での症例数を分母に、有害事象件数を分子にすることである。その他にも、試験の規模に基づいた、あるいはバラツキの大きさに反比例させたデータの重みづけをすることも可能である。
試験間の有害事象発現率が大きく異なる場合は、その旨を記載し、考えられる理由について考察すること(例:被験者集団、投与方法、有害事象データ収集法)。
有害事象の記載内容は、個別総括報告書と同じとすること(ICH E3)。多くの試験を併合する場合は、標準的な用語を用いて表現し、類義語は基本語(preferred term)に統一することが重要である。そのためMedDRA用語集(ICH M1)等一つの標準的辞書を用いること。MedDRAが完全実施されるまでは、他の辞書を用いてもよい。その場合、使用した用語集を示すこと。有害事象は、基本語ごとや定義された事象のグループごとに頻度を示すこと。
治療変更(投与中止、用量変更、治療の追加)をもたらした有害事象にどのようなものがあったか検討することは、有害事象の臨床的重要性を評価するのに有用である。これらの有害事象の発現率を有害事象一覧表に含めてもよいし、別表としてもよい。試験別にトータルの中止率を示すことは有用である。しかし、別表を用い、投与中止に至った特定の有害事象を明記することも重要である。有害事象は、器官別にグループ化し、頻度の高い順に並べること。
2.7.4.2 Adverse Events
2.7.4.2.1 Analysis of Adverse Events
Data on the frequency of adverse events should be described in text and tables. Text should appear in the appropriate subsections of Section 2.7.4.2.1 and the tables that are not embedded in the text should be placed in the section 2.7.4.7 Appendix.
All adverse events occurring or worsening after treatment has begun (“treatment emergent signs and symptoms,” those adverse events not seen at baseline and those that worsened even if present at baseline) should be summarised in tables listing each event, the number of subjects in whom the event occurred and the frequency of occurrence in subjects treated with the drug under investigation, with comparator drugs, and with placebo. Such tables could also present results for each dose and could be modified to show, e.g., adverse event rates by severity, by time from onset of therapy, or by assessment of causality.
When most of the relevant safety data are derived from a small number of studies (e.g., one or two studies), or when very different study subject populations were enrolled in the studies that were performed, presentation of data by study will often be appropriate. When the relevant exposure data is not concentrated in a small number of studies, however, grouping the studies and pooling the results to improve precision of estimates and sensitivity to differences should generally be considered.
While often useful, pooling of safety data across studies should be approached with caution because in some cases interpretation can be difficult, and it can obscure real differences. In cases where differences are apparent, it is more appropriate to present the data by study. The following issues should be considered:
- it is most appropriate to combine data from studies that are of similar design, e.g., similar in dose, duration, methods of determining adverse events, and population.
- if the incidence for a particular adverse event differs substantially across the individual studies in a pool, the pooled estimate is less informative.
- any study with an unusual adverse event pattern should be presented separately.
- the appropriate extent of analysis depends on the seriousness of the adverse event and the strength of evidence of drug causation. Differences in rates of drugrelated, serious events or events leading to discontinuation or dosage change deserve more investigation, whereas rates of other adverse events do not merit elaborate analysis.
- examination of which subjects experience extreme laboratory value abnormalities (“outliers”) may be useful in identifying subgroups of individuals who are at particular risk for certain adverse events.
Groups of studies that could be used in pooled safety analyses include:
- all controlled studies or subsets of controlled studies, such as all placebocontrolled studies, studies with any positive control, studies with a particular positive control, or studies of particular indications (and thus carried out in different populations). These groupings are considered the best source of information about the more common adverse events and can distinguish drugrelated events from spontaneous events. Rates in control and treatment groups should be compared.
- all studies, excluding short-term studies in healthy subjects. This grouping is most useful for evaluating rarer events.
- all studies using a particular dose route or regimen, or a particular concomitant therapy.
- studies in which adverse event reports are elicited by checklist or direct questioning, or studies in which events are volunteered.
- pools of studies by region.
It is almost always useful to carry out the first two groupings; the others chosen would vary from drug to drug and should be influenced by inspection of individual study results. Whatever methods are used, it should be recognised that, as for results of single studies, any numerical rate is often only a rough approximation of reality.
When a decision is made to pool data from several studies, the rationale for selecting the method used for pooling should be described. It is common to combine the numerator events and the denominators for the selected studies. Other methods for pooling results across studies are available, e.g., weighting data from studies on the basis of study size or inversely to their variance.
If substantial differences are seen between clinical trials in the rates of adverse events, these differences should be noted and possible reasons should be discussed (e.g., relevant differences in study populations, in dose administration, or in methods of collecting adverse event data).
Adverse events should be described as shown in the individual study report (ICH E3). In combining data from many studies, it is important to use standardised terms to describe events and collect synonymous terms under a single preferred term. This can be done with a standard dictionary, and the MedDRA terminology (ICH M1 guideline) should be used. Until MedDRA can be fully implemented, other dictionaries can be used, but should be specified. Frequencies should be presented for preferred terms and for appropriately defined groupings.
Examination of which adverse events led to change in therapy (discontinuation of drug use, change in dose, need for added therapy) can help in assessing the clinical importance of adverse events. These rates can be added to the adverse event rate tables, or can be presented in separate tables. Overall discontinuation rates by study may be useful but it is also important to specify the particular adverse events leading to discontinuation in a separate table. The preferred terms should be grouped by body system and arranged by decreasing frequency.
2.7.4.2.1.1 比較的よく見られる有害事象
有害事象発現率を表形式でまとめ(第2.7.4項の付録を参照)、被験薬群と対照薬群における発現率を比較すること。もし重症度分類と因果関係分類がなされていれば、表中に併記すると、治療群間の比較検討がし易く便利である。因果関係分類を示していても、データの中には(因果関係の有無にかかわらず)全ての有害事象を含めることに注意すること。なぜなら、因果関係の評価は本質的に主観的なものであり、実際には関係があるにもかかわらず予期されていない有害事象を除外していることがあるからである。さらに、個々の試験における被験薬群と対照薬群との間で有害事象発現率を比較し本項で要約すること。特定の試験を選択し、有害事象発現率を表としてまとめると有用であることが多い(第2.7.4項の付録、表2.7.4.4を参照)。
当該医薬品と関連すると思われ、比較的よく見られる有害事象(用量反応性があるもの、被験薬とプラセボとの間で発現率が明らかに異なるもの)を、以下の因子との関連について検討しつつ厳密に評価することは一般的に有用である。
- 投与量
- mg/kg又はmg/m2で示される投与量
- 投与方法
- 投与期間
- 総投与量
- 年齢、性別、人種等の人口統計学的特性
- 併用医薬品
- 腎機能等ベースライン特性
- 有効性の結果
- データがあれば、薬物の濃度
これら薬剤と関連のある有害事象を、発現時期や持続時間との関連で検討した結果の要約もまた有用と考えられる。
ただし、上記の各因子それぞれと有害事象との関連について、一般的に、厳密な統計的評価を行う必要はない。データをまとめた時点や検討した時点で既に人口統計学的特性、その他のベースライン特性に対し意味ある関連性がないことが明らかな場合もある。そのような場合、特定の因子についてさらに解析を行うことは不要であるし、解析をしてもその結果をこの項に記載する必要はない。また、安全性の解析結果が広範なもので、その詳細を示すことができない場合には、第5部第5.3.5.3項に示し、本項ではその要約を示せばよい。
内容によっては、有害事象の発現率をそのまま記述するよりも、生命表法や類似の解析方法で報告する方が望ましいこともある。
2.7.4.2.1.1 Common Adverse Events
Tabular displays of adverse event rates (see the section 2.7.4.7 Appendix) should be used to compare rates in treatment and control groups. For this analysis it may be helpful to combine the event severity categories and the causality categories, if they are used, leading to a simpler side-by-side comparison of treatment groups. It should be noted that while causality categories may be reported, if used, the presentation of the data should include total adverse events (whether deemed related or unrelated to treatment); evaluations of causality are inherently subjective and may exclude unexpected adverse events that are in fact treatment related. Additionally, comparisons of rates of adverse events between treatment and control groups in individual trials should be summarised here. It is often useful to tabulate rates in selected trials (see example table 2.7.4.4, in the Section 2.7.4.7 Appendix).
It is usually useful to examine more closely the more common adverse events that seem to be drug related (e.g., those that show that a dose response and/or a clear difference between drug and placebo rates) for relationship to relevant factors, including:
- dosage;
- mg/kg or mg/m2 dose;
- dose regimen;
- duration of treatment;
- total dose;
- demographic characteristics such as age, sex, race;
- concomitant medication use;
- other baseline features such as renal status;
- efficacy outcomes;
- drug concentration, where available.
It may also be useful to summarise the results of examination of time of onset and duration for these drug-related events.
Rigorous statistical evaluations of the possible relationship of specific adverse events to each of the above factors are often unnecessary. It may be apparent from initial display and inspection of the data that there is no evidence of a significant relationship to demographic or other baseline features. In that case, no further analysis of these particular factors is needed. Further, it is not necessary that all such analyses be presented in this report. When the safety analyses are too extensive to be presented in detail in this report, they may be presented in a separate report in Module 5, section 5.3.5.3, and summarised here.
Under certain circumstances, life table or similar analyses may be more informative than reporting of crude adverse event rates.
2.7.4.2.1.2 死亡
治験中の死亡例は、すべて第2.7.4項付録中の表に列記すること(試験終了後短期間での死亡、例えば中止後30日以内又は治験プロトコールに規定された期間内の死亡、それ以後の事例ながら試験中の何らかの経過(process)に起因したと考えられる死亡例)。ただし、進行癌等の死亡率の高い集団での試験や疾患に起因する死亡率が主要評価項目となっている試験において、プロトコールに規定された疾患に関連し、治験薬に関連しない死亡例(これらの死亡例は、ICH E3治験総括報告書に報告することとされている)はこの一覧表から除外すること。これらの死亡についても、群間に予想外のパターンがなかったかどうかを検討し、新たな差異が観察された場合には解析を追加すること。死亡例はそれぞれについて評価し、個々の試験及び全体における総死亡率と原因別死亡率について分析すること。第2.7.4.2.1.1項に示した因子との関係も考慮すること。死亡原因を特定することは難しいかもしれないが、比較的解釈しやすいものもある。対象となる患者集団において予想される原因による死亡例(例:狭心症患者集団における心臓発作及び突然死)一つ一つに情報価値はないと考えられるかも知れないが、例え1症例であってもそれがQT時間延長を伴う不整脈、再生不良性貧血、肝障害によるもの等であれば重要情報となる。死亡原因を合併症に関連付けるにあたっては、特別の注意を払うこと。
2.7.4.2.1.2 Deaths
A table in the Section 2.7.4.7 Appendix should list all deaths occurring while on study (including deaths that occurred shortly following treatment termination, e.g., within 30 days or as specified in the study protocol, as well as all other deaths that occurred later but may have resulted from a process that began during studies). Only deaths that are clearly disease-related per protocol definitions and not related to the investigational product, either in studies of conditions with high mortality such as advanced cancer or in studies where mortality from disease is a primary study endpoint, should be excepted from this listing (it is assumed, however, that these deaths would still be reported in the individual ICH E3 study reports). Even these deaths should be examined for any unexpected patterns between study arms, and further analysed if unexplained differences are observed. Deaths should be examined individually and analysed on the basis of rates in individual trials and appropriate pools of trials, considering both total mortality and cause-specific deaths. Potential relationships to the factors listed in Section 2.7.4.2.1.1 should also be considered. Although causespecific mortality can be difficult to determine, some deaths are relatively easy to interpret. Thus deaths due to causes expected in the patient population (heart attacks and sudden death in an angina population) are individually not considered to be informative, but even one death due to a QT interval prolongation-associated arrhythmia, aplastic anaemia, or liver injury may be informative. Special caution is appropriate before an unusual death is attributed to concomitant illness.
2.7.4.2.1.3 その他の重篤な有害事象
本項に、全ての重篤な有害事象(死亡ではないが、時間的に死亡に関連する、又は死亡に先行する重篤な有害事象を含む)を要約すること。また、被験薬を中止した後に発現した重篤な有害事象も含めること。その他、ICH E2A定義により重篤な有害事象と思われた臨床検査値異常、異常なバイタルサイン、異常な身体的観察項目を含めること。重篤な有害事象については経時的頻度を検討すること。特に、長期に亘って使用される可能性のある医薬品については検討を要する。また、第2.7.4.2.1.1項に示した因子との関連性も考察すること。
2.7.4.2.1.3 Other Serious Adverse Events
Summaries of all serious adverse events (other than death but including the serious adverse events temporally associated with or preceding the deaths) should be displayed. Serious adverse events that occurred after the drug use was discontinued should be included in this section. The display should include major laboratory abnormalities, abnormal vital signs, and abnormal physical observations that are considered serious adverse events using the ICH E2A definitions. Results of analyses or assessments of serious adverse events across studies should be presented. Serious events should be examined for frequency over time, particularly for drugs that may be used chronically. Potential relationships to the factors listed in Section 2.7.4.2.1.1 should also be considered.
2.7.4.2.1.4 その他の重要な有害事象
顕著な血液学的異常、又はその他の臨床検査値異常(重篤という定義を満たすものを除く)、及び何らかの処置(治験薬の中止、減量又は重要な併用療法の追加等)を必要とした全ての事象を示すこと。
治験薬の投与中止をもたらす有害事象は重要な安全性上の関心事であり、次の二つの理由から安全性解析には特別の注意を払う必要がある。第一に、(薬理活性に基づき)予期されていたものであれ、中止(又は他剤に変更)する必要性があったということは、患者や医師がその有害事象の重症度と重要性をいかに感じたかを示している。第二に、投薬中止は、まだ因果関係が認識されていない治験薬と関連する有害事象であることを意味する場合がある。中止に至る有害事象は、当初その因果関係が認識されていなくとも、また、偶発症と考えられる場合でも、治験薬との関連性を否定できないと考えること。中止について、その理由を検討し、中止率を試験間並びに治験薬群とプラセボ群、実薬対照群との間で比較すること。さらに、第2.7.4.2.1.1項に示した各因子と関連があるかどうかデータを検討すること。
2.7.4.2.1.4 Other Significant Adverse Events
Marked haematologic and other laboratory abnormalities (other than those meeting the definition of serious) and any events that led to a substantial intervention (premature discontinuation of study drug, dose reduction, or substantial additional concomitant therapy), other than those reported as serious adverse events, should be displayed.
Events that led to premature discontinuation of study drug represent an important safety concern and deserve particular attention in the analysis of drug safety for two reasons. First, even for expected events (based on pharmacologic activity), the need to discontinue (or otherwise alter) treatment reflects the severity and perceived importance of the event to patient and physician. Second, discontinuation may represent a drug-related event not yet recognised as drug related. Adverse events leading to treatment discontinuation should be considered possibly drug-related even if this was not recognised initially and even if the event was thought to represent intercurrent illness. Reasons for premature treatment discontinuations should be discussed and rates of discontinuations should be compared across studies and compared with those for placebo and/or active control treatment. In addition, the study data should be examined for any potential relationships to the factors listed in Section 2.7.4.2.1.1.
2.7.4.2.1.5 器官別又は症候群別有害事象の解析
死亡、その他の重篤な、又は重要な有害事象の因果関係や危険因子についての評価は、それらの事象が高頻度で起こらないため、多くの場合、簡単でない。その結果、相互に関連性のある事象を、例え重要性の低い病態生理学的事象であれ、一つのグループとしてまとめ検討することが、安全性の特徴を理解する上で有用なこともある。例えば、突然死が1例見られた時、治療との因果関係があるかどうかは、失神、動悸や無症候性不整脈の発現状況と共に考察することにより明らかになることもある。
このように、有害事象を器官別にまとめれば、臨床検査値異常を含め諸事象との関連性を検討することもできる。器官別のまとめは、第2.7.4.2.1.5項中の第2.7.4.2.1.5.1項、第2.7.4.2.1.5.2項等のサブセクションとして記載し、表題には当該器官名を付すこと。器官のまとめ方や有害事象のグループ化は、有害事象データが理解しやすいよう配慮すること。有害事象が症候群(例:インフルエンザ様症候群、サイトカイン放出症候群)として現れる場合には、器官別ではなく、症候群別用に第2.7.4.2.1.5項のサブセクションをおこしてもよい。
同じデータや要約は、第2.7.4.2.1項のサブセクションに一度記載すればよく、繰り返さないこと。必要があれば相互参照すること。
2.7.4.2.1.5 Analysis of Adverse Events by Organ System or Syndrome
Assessment of the causality of, and risk factors for, deaths, other serious events, and other significant events is often complicated by the fact that they are uncommon. As a result, consideration of related events as a group, including less important events of potentially related pathophysiology, may be of critical value in understanding the safety profile. For example, the relationship to treatment of an isolated sudden death may become much clearer when considered in the context of cases of syncope, palpitations, and asymptomatic arrhythmias.
It is thus generally useful to summarise adverse events by organ system so that they may be considered in the context of potentially related events including laboratory abnormalities. Such presentations of adverse events by organ system should be placed in subsections of section 2.7.4.2.1.5, labelled as 2.7.4.2.1.5.1, 2.7.4.2.1.5.2, etc., and titled by the organ system under consideration. The list of organ systems to be addressed and the approach to grouping certain events should be selected as appropriate to best present the adverse event data for the medicinal product. If some adverse events tend to occur in syndromes (e.g., influenza-like syndrome, cytokine release syndrome), the sponsor may choose to create some subsections of 2.7.4.2.1.5 for syndromes rather than organ systems.
The same data and summarisations should generally not be repeated in more than one subsection of Section 2.7.4.2.1. Instead, a summary presentation may be placed in one subsection and cross-referenced as needed in the other.
2.7.4.2.2 個別有害事象の文章による説明
死亡、その他の重篤な有害事象、他の重要な有害事象は、臨床的に重要な関心事であるため(ICH E3、治験総括報告書に記載)、事例それぞれの記述が申請資料中のどこにあるかを、審査担当者の利便のため、本項にて示すこと。治験総括報告書が作成されている場合、事例報告そのものは、その一部として存在するはずである。もしも作成されていない場合(例:安全性解析のために全てのオープン試験を併合しており、個々の報告書が作成されていない)、記述は第5部第5.3.5.3項に示してもよい。したがって、ごく簡単にでも説明しておかなくては当該医薬品の評価に支障があると考えられない限り、本項には含めないこと。
2.7.4.2.2 Narratives
The locations in the application of individual narratives of patient deaths, other serious adverse events, and other significant adverse events deemed to be of special interest because of clinical importance (as described in ICH E3 individual study reports) should be referenced here for the convenience of the reviewer. The narratives themselves should be a part of the individual study reports, if there is such a report. In cases where there is no individual study report (e.g., if many open studies are pooled as part of a safety analysis and are not individually described), narratives can be placed in Module 5, Section 5.3.5.3. Narratives should not be included here, unless an abbreviated narrative of particular events is considered critical to the summary assessment of the drug.
2.7.4.3 臨床検査値の評価
本項では、被験薬の使用に伴う臨床検査値の変動パターンを要約すること。顕著な異常や何らかの処置を要したものは第2.7.4.2.1.3項又は第2.7.4.2.1.4項に報告すること。もし、それらのデータを本項に記載する場合は、内容が重複していることを示すこと。臨床検査値の評価は、得られた結果そのものから行いうるが、通常、以下に列記する解析も実施し記載すること。各解析は、試験規模を考慮しつつ、被験薬群と対照薬群とで比較すること。各検査項目に対し正常範囲を示すこと(ICH E3)。可能であれば、臨床検査値は国際標準単位で示すこと。
全ての試験を通して、重要な臨床検査値の変動に関し簡単に概括すること。臨床検査データとしては、血液学的検査、血液生化学的検査、尿検査、その他必要に応じて実施された検査データを含む。治験期間中の各測定点(来院時等)で得られた各パラメータは以下の3段階にて表記すること。
- 中心傾向、例、群の平均値及び中央値
- 値の範囲及び異常値を示した被験者数、又は特定の範囲(例:正常値範囲の上限の2倍、上限の5倍等。その選択について説明すること)の異常値を示した被験者数。もしも、正常範囲が異なる施設からのデータを併合する時、併合に用いた方法を記載すること。各投与群における被験者の変動の解析には適宜いろいろな方法を用いることができる(例:シフトテーブル、ICH E3を参照)。
- 中止例を含む、臨床的に重要な個々の臨床検査値異常。
これらの臨床検査値の変動についてその重要性及び医薬品との関連性を評価すること(例:投与量との関連性、薬物濃度との関連性、治療継続で消失、投与中止で消失、再投与で再発、併用療法の内容)。第2.7.4.2.1.1項で示した他の因子との関連についても検討すること。
2.7.4.3 Clinical Laboratory Evaluations
This section should describe changes in patterns of laboratory tests with drug use. Marked laboratory abnormalities and those that led to a substantial intervention should be reported in section 2.7.4.2.1.3 or 2.7.4.2.1.4. If these data are also presented in this section, this duplicate reporting should be made clear for the reviewer. The appropriate evaluations of laboratory values will in part be determined by the results seen, but, in general, the analyses described below should be provided. For each analysis, comparison of the treatment and control groups should be carried out, as appropriate and as compatible with study sizes. In addition, normal laboratory ranges should be given for each analysis (ICH E3). Where possible, laboratory values should be provided in standard international units.
A brief overview of the major changes in laboratory values across the clinical studies should be provided. Laboratory data should include haematology, clinical chemistry, urinalysis and other data as appropriate. Each parameter at each time over the course of the study (e.g., at each visit) should be described at the following three levels:
- the central tendency, i.e., the group mean and median values,
- the range of values, and the number of subjects with abnormal values or with abnormal values of a certain size (e.g. twice the upper limit of normal, 5 times the upper limit; choices should be explained). When data are pooled from centres with differences in normal laboratory ranges, the methodology used in pooling should be described. The analysis of individual subject changes by treatment group can be shown with a variety of approaches (e.g., shift tables, see ICH E3 for examples).
- individual clinically important abnormalities, including those leading to discontinuations.
The significance of the laboratory changes and the likely relation to the treatment should be assessed (e.g., by analysis of such features as relationship to dose, relation to drug concentration, disappearance on continued therapy, positive dechallenge, positive rechallenge, and the nature of concomitant therapy). Potential relationships to other factors listed in Section 2.7.4.2.1.1 should also be considered.
2.7.4.4 バイタルサイン、身体的所見及び安全性に関連する他の観察項目
全ての試験を通してバイタルサイン(例:心拍数、血圧、体温、呼吸数)、体重及び安全性に関連するその他のデータ(例:心電図、X線)を比較する方法は、基本的に前記の臨床検査値と同じ方法にて行うこと。被験薬が影響を及ぼしている証拠がある場合、用量-反応性もしくは薬物濃度-反応関係、又は患者の特性(例:疾患、人口統計学的特性、併用療法)との関係を明らかにし、この所見の臨床上の意義について記述すること。
有効性の項目として評価されない変化及び有害事象とみなされる変化に特別の注意を払うこと。QT間隔延長に関する試験等、特別な安全性上の問題を評価するために計画された試験には、特別の注意を払うこと。
2.7.4.4 Vital Signs, Physical Findings, and Other Observations Related to Safety
The manner of presenting cross-study observations and comparisons of vital signs (e.g., heart rate, blood pressure, temperature, respiratory rate), weight and other data (e.g., electrocardiograms, X-rays) related to safety should be similar to that for laboratory variables. If there is evidence of a drug effect, any dose-response or drug concentration-response relationship or relationship to individual variables (e.g., disease, demographics, concomitant therapy) should be identified and the clinical relevance of the observation described.
Particular attention should be given to changes not evaluated as efficacy variables and to those considered to be adverse events. Particular attention should be given to studies that were designed to evaluate specific safety issues, e.g., studies of QT interval prolongation.
2.7.4.5 特別な患者集団及び状況下における安全性
2.7.4.5.1 内因性要因
本項では、ICH E5にて「内因性民族的要因」と定義される人口統計学的因子や他の要因に基づいて治療及び管理を患者個々に考慮するための安全性データを要約すること。内因性要因には、年齢、性別、身長、体重、除脂肪体重、遺伝子多型、身体組成、他の疾患及び臓器機能不全が含まれる。申請適応症が小児でも考えられる場合は、小児における安全性も分析すること。これらの要因が安全性に与える影響については他項に記載されることになるが、腎疾患又は肝疾患患者等における安全性は、薬物動態、その他の情報と共に本項で要約すること。もし、高血圧、心疾患、糖尿病等の特定の合併症のある患者の例数が十分であれば、合併症が治験薬の安全性にどのような影響を与えるのか分析すること。患者部分集団における解析を行う場合は、作成した有害事象の表又は記述を相互に参照すること。
2.7.4.5 Safety in Special Groups and Situations
2.7.4.5.1 Intrinsic Factors
This section should summarise safety data pertinent to individualising therapy or patient management on the basis of demographic and other factors defined as “intrinsic ethnic factors” in ICH E5. These factors include age, sex, height, weight, lean body mass, genetic polymorphism, body composition, other illness and organ dysfunction. Safety in the paediatric population should be routinely analysed in applications for a proposed indication that occurs in children. Analysis of the impact of such factors on safety outcomes should have been presented in other sections but should be summarised here, together with pertinent PK or other information, e.g., in patients with renal or hepatic disease. If a sufficiently large number of subjects with a given co-morbid condition such as hypertension, heart disease, or diabetes, was enrolled, analyses should be carried out to assess whether the co-morbid condition affected the safety of the drug under study. Cross reference should be made to the tables or description of adverse events when analyses of such sub-groups has been carried out.
2.7.4.5.2 外因性要因
本項では、ICH E5にて「外因性民族的要因」と定義される人口統計学的因子や他の要因に基づいて治療及び管理を患者個々に考慮するための安全性データを要約すること。これらの因子は、患者をとりまく環境に関する因子である。例としては、医療環境、他剤の使用状況(第2.7.4.5.3項、薬物相互作用を参照)、喫煙、飲酒、食事の習慣等が挙げられる。
例えば、代謝プロフィール、試験結果、市販後調査又は類薬情報等により飲酒と治験薬との相互作用が示唆される場合、それらの内容を本項に示すこと。
2.7.4.5.2 Extrinsic Factors
This section should summarise safety data pertinent to individualising therapy or patient management on the basis of factors defined as “extrinsic ethnic factors” in ICH E5. These are factors associated with the patient environment. Examples are the medical environment, use of other drugs (see 2.7.4.5.3, Drug Interactions), use of tobacco, use of alcohol, and food habits.
For example, if a potential interaction with alcohol is suggested by the metabolic profile, by the results of studies, by post-marketing experience, or by information on similar drugs, information should be provided here.
2.7.4.5.3 薬物相互作用
薬物-薬物又は薬物-食事相互作用に関する試験は、CTD臨床薬理試験の要約の項(第2.7.2項)に要約すること。これらの相互作用が安全性に与える影響については、薬物動態、薬力学や臨床的観察に基づいて分析し本項で要約すること。さらに、他の治療との併用時に観察された有害事象プロフィールの変化、リスクに結びつきそうな血中濃度の変化、被験薬の薬効の変化等を本項で示すこと。
2.7.4.5.3 Drug Interactions
Studies on potential drug-drug or drug-food interactions should be summarised in the Summary of Clinical Pharmacology Studies section of the CTD (Section 2.7.2). The potential impact on safety of such interactions should be summarised here, based on PK, PD, or clinical observations. Any observed changes in the adverse event profile, changes in blood levels thought to be associated with risk, or changes in drug effects associated with other therapy should be presented here.
2.7.4.5.4 妊娠及び授乳時の使用
開発中及び他の情報源から得られた妊娠及び授乳時の安全性に関する情報を本項で要約すること。
2.7.4.5.4 Use in Pregnancy and Lactation
Any information on safety of use during pregnancy or breast-feeding that becomes available during clinical development or from other sources should be summarised here.
2.7.4.5.5 過量投与
過量投与に関連すると思われる臨床症状、臨床検査値所見、対症療法、解毒薬の使用等のデータがあれば、全ての情報を要約し考察すること。また、過量投与に対する解毒薬や透析の有効性も、データがあれば記載すること。
2.7.4.5.5 Overdose
All available clinical information relevant to overdose, including signs/symptoms, laboratory findings, and therapeutic measures/treatments and antidotes (if available) should be summarised and discussed. Information on the efficacy of specific antidotes and dialysis should be provided if available.
2.7.4.5.6 薬物乱用
ヒト及び動物における新薬の依存性に関する試験や情報を要約し、「非臨床概要」を参照すること。特に依存性を生じやすい患者集団を明らかにすること。
2.7.4.5.6 Drug Abuse
Any relevant studies/information regarding the investigation of the dependence potential of a new therapeutic agent in animals and in humans should be summarised and cross-referenced to the nonclinical summary. Particularly susceptible patient populations should be identified.
2.7.4.5.7 離脱症状及び反跳現象
当該被験薬の反跳作用に関する試験結果及び情報を要約すること。二重盲検試験や実薬投与の中止後に何らかの事象が発現したり、重症度を増したりした場合、それが投与中止によるものかどうか検討すること。離脱症状、反跳現象を評価するよう計画された試験には特に重視すること。
耐薬性に関するデータを「臨床的有効性の概要」の第2.7.3.5項に要約すること。
2.7.4.5.7 Withdrawal and Rebound
Any information or study results pertinent to rebound effects should be summarised. Events that occur, or increase in severity, after discontinuation of double-blind or active study medication should be examined to see if they are the result of withdrawal of the study medication. Particular emphasis should be given to studies designed to evaluate withdrawal and/or rebound.
Data concerning tolerance should be summarised under section 2.7.3.5 in the Summary of Clinical Efficacy.
2.7.4.5.8 自動車運転及び機械操作に対する影響又は精神機能の障害
五感、協調運動、その他の障害に基づく自動車運転や機械操作能の低下、精神機能の障害等に関する安全性データを要約すること。これには安全性モニタリングで報告された関連する有害事象(例:眠気)及び自動車の運転や機械操作能力に及ぼす影響、精神機能障害に関する特別な試験が含まれる。
2.7.4.5.8 Effects on Ability to Drive or Operate Machinery or Impairment of Mental Ability
Safety data related to any impairment in the senses, co-ordination, or other factor that would result in diminished ability to drive a vehicle or operate machinery or that would impair mental ability should be summarised. This includes relevant adverse effects reported in safety monitoring (e.g., drowsiness) and specific studies concerning effects on ability to drive or operate machinery or impairment of mental ability.
2.7.4.6 市販後データ
当該医薬品が既に販売されている場合、申請者が入手できる全ての市販後データ(安全性定期報告を含む既発表又は未発表資料)を要約すること。定期安全性情報は第5部に含めてもよい。
曝露されたと推測される患者数を、適応症、投与量、投与経路、投与期間、治験実施地域等適切に分類し記載すること。そして、患者数を推定するために用いた方法を記載すること。人口統計学的特性の評価があれば、それを示すこと。
市販後、報告された重篤な有害事象を表としてまとめること。もし、重篤な薬物相互作用の可能性を示唆するものがあれば含めること。
市販後、種々の部分集団にて得られたいかなる知見も記載すること。
2.7.4.6 Post-marketing Data
If the drug has already been marketed, all relevant post-marketing data available to the applicant (published and unpublished, including periodic safety update reports if available) should be summarised. The periodic safety update reports can be included in Module 5.
Details of the number of subjects estimated to have been exposed should be provided and categorised, as appropriate, by indication, dosage, route, treatment duration, and geographic location. The methodology used to estimate the number of subjects exposed should be described. If estimates of the demographic details are available from any source, these should be provided.
A tabulation of serious events reported after the drug is marketed should be provided, including any potentially serious drug interactions.
Any post-marketing findings in subgroups should be described.
2.7.4.7 付録
安全性評価のための全ての試験について、重要な結果と、特に、添付文書の内容を裏付ける結果を表形式にまとめること。
文書が読みやすくなるのであれば、図や表を適切に文章中に挿入すること。大きな表は、本項の最後に付録として添付することができる。
作表例を以下にいくつか例示するが、臨床概要では、それぞれの医薬品、医薬品分類及び適応症に応じた作表や作図が、通常、必要となるはずである。
第2.7.4項に含める表の種類・内容については、本ガイドラインの第2.7.4.2.1項、第2.7.4.2.2.3項及び第2.7.4.3項も参照すること。
表2.7.4.1 平均1日投与量及び投与期間別曝露
表2.7.4.2 比較対照試験における被験者の人口統計学的特性
表2.7.4.3 併合したプラセボ及び実薬対照比較試験における有害事象発現率
表2.7.4.4 選択した特定の試験における有害事象発現率
表2.7.4.5 試験別の中止例:比較対照試験
表2.7.4.6 死亡例一覧
2.7.4.7 Appendix
Tabular presentations should be provided that summarise the important results from all studies pertinent to the evaluation of safety and particularly to support product labelling.
Tables and figures should be embedded in the text of the appropriate sections when that enhances the readability of the document. Lengthy tables can be provided in the appendix at the end of the Section.
A few illustrative tables are provided, but a clinical summary will routinely need tables and figures that have been developed for the particular drug, drug class, and clinical indication(s).
See sections 2.7.4.2.1, 2.7.4.2.2.3, and 2.7.4.3 of this guidance for additional discussion regarding the content of section 2.7.4 tables.
Table 2.7.4.1 Study Subject Drug Exposure by Mean Daily Dose and Duration of Exposure
Table 2.7.4.2 Demographic Profile of Patients in Controlled Trials
Table 2.7.4.3 Incidence of Adverse Events in Pooled Placebo and Active Controlled Trials
Table 2.7.4.4 Incidence of Adverse Events in the Largest Trials
Table 2.7.4.5 Patient Withdrawals by Study: Controlled Trials
Table 2.7.4.6 Listing of Deaths
2.7.5 参考文献
臨床概要にて引用した参考文献を列記すること。重要文献は、全てコピーし第5部第5.4項に添付すること。文献リストには、コピー添付の有無を示しておくこと。また、コピーを添付しない文献は、要求に基づき提供できるようにしておくこと。
2.7.5 Literature References
A list of references cited in the Clinical Summary should be provided. Copies of all important references should be provided in Module 5, Section 5.4. The reference list should indicate which references are available in Module 5, Section 5.4. All references that have not been provided should be available upon request.
2.7.6 個々の試験のまとめ
ICH E3ガイドライン(「治験の総括報告書の構成と内容」)では、治験総括報告書にはシノプシス(概要)を含めることを提案し、また、その様式も例示されている。
本項に、臨床試験一覧表と個々の試験の概要を、第5部の試験報告書の添付順序と同じ順序で記載すること。
シノプシスは、試験ごとに一つ作成し、全ての地域で共通使用すること、また、本項及び第5部(モジュール5)の臨床試験報告書の一部として使用することになっている。長さは通常3ページ以下であるが、複雑かつ重要な試験の場合は長くてもよい(例:10ページ)。概要の理解のため必要であれば、表や図を加えること。
2.7.6 Synopses of Individual Studies
The ICH E3 guideline (Structure and Content of Clinical Study Reports) suggests inclusion of a study synopsis with each clinical study report, and provides one example of a format for such synopses.
This section should include the table entitled Listing of Clinical Studies, described in guidance for Module 5, followed by all individual study synopses organised in the same sequence as the study reports in Module 5.
It is expected that one synopsis will be prepared per study for use in all regions, and that the same synopsis will be included in this section and as part of the clinical study report in Module 5. The length of a synopsis will usually be up to 3 pages, but a synopsis for a more complex and important study may be longer, e.g. 10 pages. Within the individual synopsis, tables and figures should be used as appropriate to aid clarity.





