

CSRの文書内の整合性
Information Consistency Across CSR
薬審第335号:治験の総括報告書の構成と内容

ICH-E3: Structure and Content of Clinical Study Reports

CSRの文書内の整合性(当社での代表的な確認項目の一部を記載)
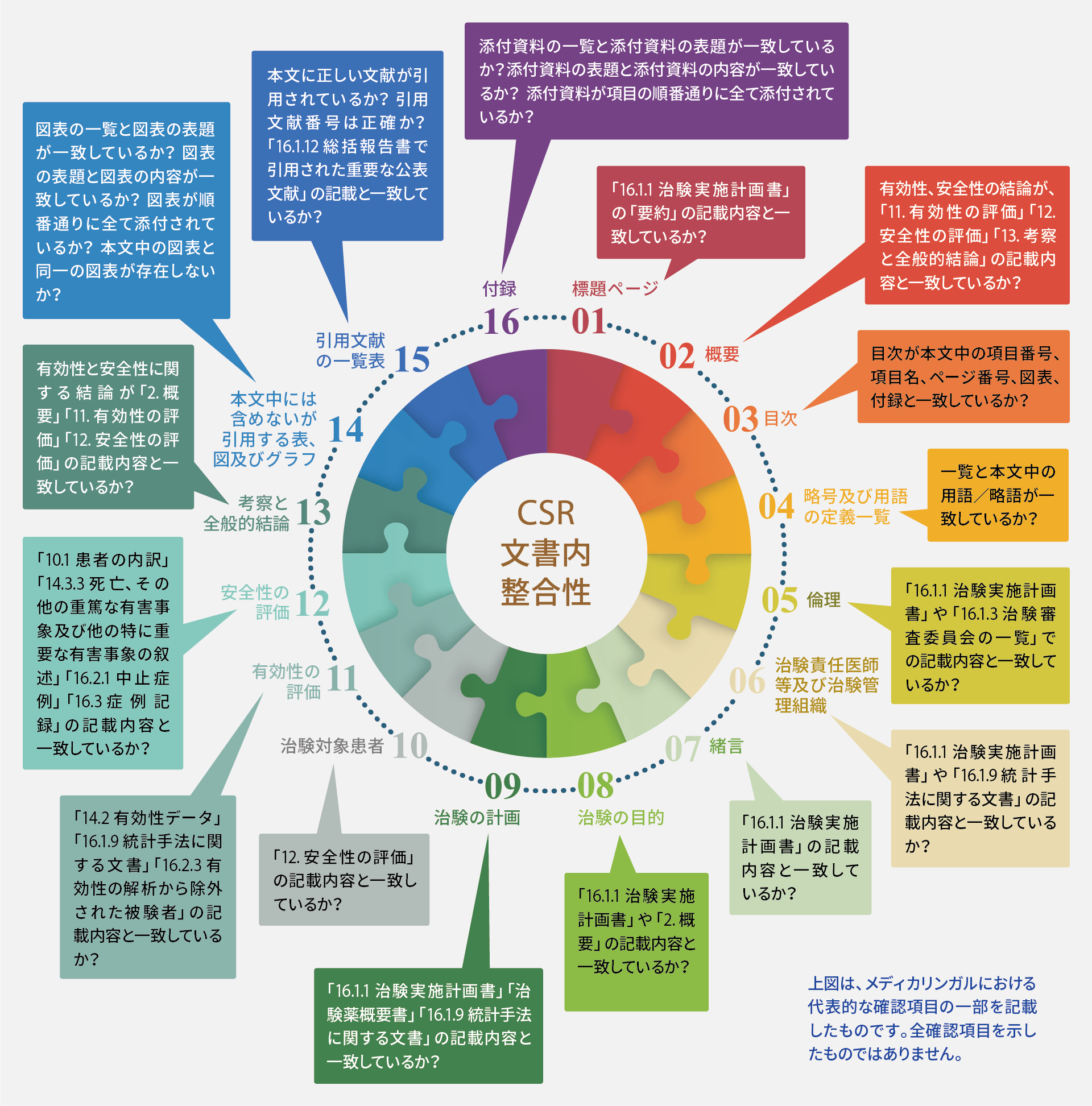
Information Consistency Across CSR (Shows only typical examples at MedicaLingual work)
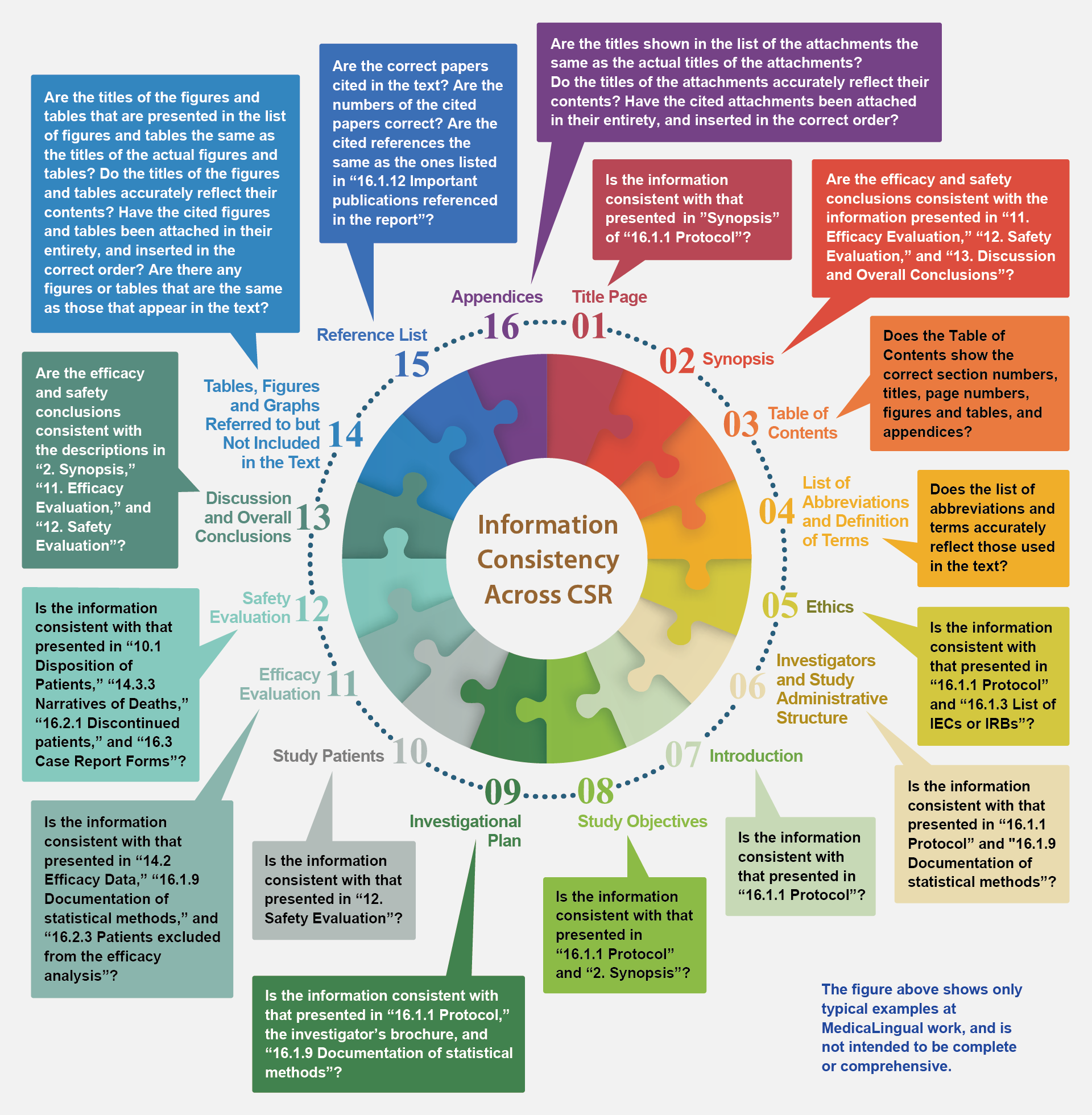
CSR記載内容の整合性及び科学的妥当性の確認
Review of CSRs for Consistency and Scientific Appropriateness

当社では、CSRの文書内の整合性、及びCSRと他の文書間の整合性の確認作業を行っています。
また、英語ネイティブのメディカルライターによる英文校正を含むCSRの内容確認や、医学専門家によるCSRの科学的妥当性の確認作業も行っております。
MedicaLingual checks consistency of information both across the CSR and between the CSR and other documents.
MedicaLingual can also arrange for native English medical writer to review English-language CSRs for content as well as grammar and style, and for medical experts to review CSRs for scientific appropriateness.
治験総括報告書の翻訳と作成
Translation & Preparation of CSR
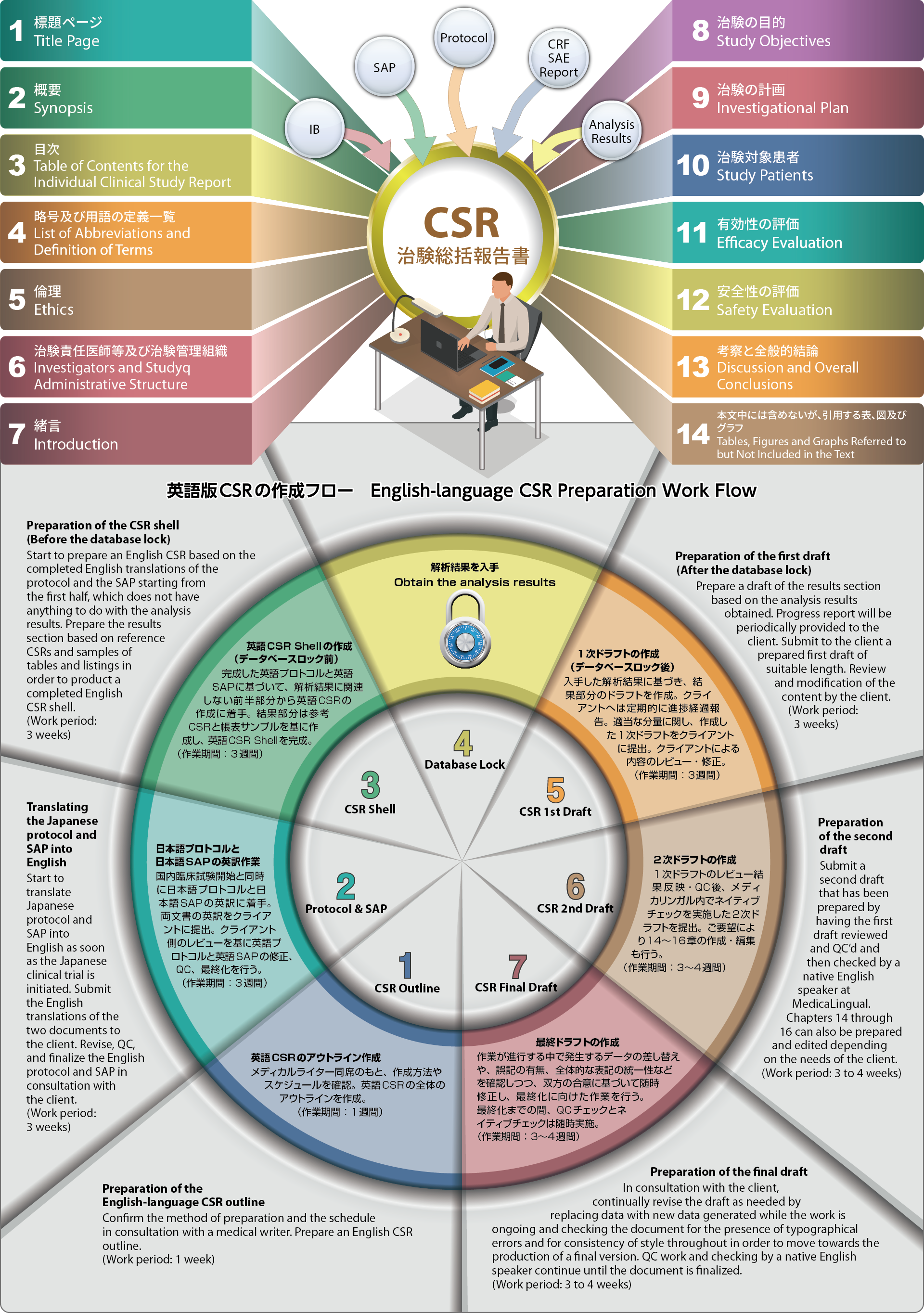
当社では医薬品申請文書の翻訳とメディカルライティングを同時並行で行うことができます。また、お客様からご提供いただいたプロトコール、SAP、IB、解析結果、CRF、SAE Report等を基に、日本語または英語でのCSRを作成することも可能です。
また、当社では国内臨床試験と同時並行で英語版CSRを作成することができます。国内臨床試験開始と同時に日本語版のプロトコール、SAP、IBの英訳に着手し、完成した英語版プロトコール、SAP、IBを基に、データベースロック後の解析結果から英語版CSRを作成します。
CSRの翻訳及びメディカルライティング業務においては、当社からお客様への初回納品後に、以下の一連のプロセスを経て最終化に至ります。
①クライアント側での複数回にわたるレビューと修正
②クライアントからのフィードバックを受けての当社側での修正(クライアント側での必要に応じ複数回実施)
③当社側での英語ネイティブチェック(英語メディカルライティングの場合)
④クライアント側で確認された最終ドラフトに対する、当社側でのQC点検及びQC点検後の最終版の作成(CSR規定テンプレート編集を含む)
⑤最終版納品後、QC点検証跡、品質管理証明書、翻訳証明書を提出

MedicaLingual is able to provide our clients with simultaneous approval application translation and medical writing services and to prepare Japanese-language or English-language CSRs based on protocols, SAPs, IBs, analysis results, CRFs, SAE reports, etc. provided by our clients.
MedicaLingual can also prepare English-language CSRs while the Japanese clinical studies are ongoing. MedicaLingual starts translating the Japanese protocol, SAP, and IB into English as soon as the Japanese clinical trial has been initiated, and prepares the English-language CSR based on the completed English-language translations of the protocol, SAP, and IB and on the analysis results obtained after the database lock.
Our CSR translation and medical writing services cover the following series of processes until the finalization after the first delivery to the client from us:
After an extensive review and revision process in which the document is reviewed by the client, the document is revised by MedicaLingual based on the feedback received multiple times, depending on the needs of the client.
It may then also be reviewed by native English speakers at MedicaLingual if it is written in English and subjected to QC inspections by MedicaLingual of the final draft confirmed by the client, followed by the preparation of a QC’d finished version (including formatting for use in specified CSR templates) at MedicaLingual.
MedicaLingual presents the client with QC inspection traceability assurance, a quality control certificate or a translation certificate for each final deliverable.

総括報告書と実施計画書間の整合性(参照すべき実施計画書の主な項目)
Information Consistency Between the CSR and the Protocol (Main sections in the Protocol that should be referenced)
| 治験の総括報告書の構成と内容(薬審第335号) | 主な参照項目:電子的に構造化・調和された臨床試験実施計画書(ICH-M11 ドラフト版) | |
| 1 | 標題ページ | 1 試験実施計画書の要約 |
| 2 | 概要 | 1 試験実施計画書の要約 |
| 3 | 目次 | |
| 4 | 略号及び用語の定義一覧 | 14 付録:用語集 |
| 5 | 倫理 | 10 一般的留意事項:規制、倫理及び試験管理 |
| 5.1 | 治験審査委員会(IRB) | 10.2 委員会 |
| 5.2 | 治験の倫理的実施 | 10.1 規制上及び倫理的な配慮 |
| 5.3 | 患者への情報及び同意 | 10.3 説明・同意取得手順 |
| 6 | 治験責任医師等及び治験管理組織 | |
| 7 | 緒言 | 2 緒言 |
| 8 | 治験の目的 | 3 試験の目的、評価項目及び estimand |
| 9 | 治験の計画 | 4 試験デザイン |
| 9.1 | 治験の全般的デザイン及び計画―記述 | 4.1 試験デザインの詳細 |
| 9.2 | 対照群の選択を含む治験デザインについての考察 | 4.2 試験デザインの設定根拠 |
| 9.3 | 治験対象集団の選択 | 5.1 試験対象集団の選択 |
| 9.3.1 | 組み入れ基準 | 5.3 選択基準 |
| 9.3.2 | 除外基準 | 5.4 除外基準 |
| 9.3.3 | 患者の治療又は評価の打ち切り | 7 試験介入の中止及び参加者の試験中止 |
| 9.4 | 治療法 | 6 試験介入及び併用療法 |
| 9.4.1 | 治療法 | 6.1 試験介入の詳細 |
| 9.4.2 | 治験薬の同定 | 6.5 調製、取扱い、保管及び管理 |
| 9.4.3 | 治療群への患者の割付け方法 | 6.6 参加者の割付け、ランダム化及び盲検化 |
| 9.4.4 | 治験における用量の選択 | 6.3 用法及び用量 |
| 9.4.5 | 各患者の用量の選択及び投与時期 | 6.3.1 試験介入の用量調節 |
| 9.4.6 | 盲検化 | 6.6.3 盲検化及び盲検解除 |
| 9.4.7 | 前治療及び併用療法 | 6.8 併用療法 |
| 9.4.8 | 治療方法の遵守 | 6.7 試験介入の遵守 |
| 9.5 | 有効性及び安全性の項目 | 8 試験の評価及び手順 |
| 9.5.1 | 有効性及び安全性の評価項目及びフローチャート | 8.2 有効性評価手順;8.3 安全性評価手順 |
| 9.5.2 | 測定項目の適切性 | 4.2 試験デザインの設定根拠 |
| 9.5.3 | 有効性の主要評価項目 | 8.2 有効性評価手順 |
| 9.5.4 | 薬物濃度の測定 | 8.7 薬物動態 |
| 9.6 | データの品質保証 | 11.2 データの品質保証 |
| 9.7 | 治験実施計画書で計画された統計手法及び症例数の決定 | 9 統計学的事項 |
| 9.7.1 | 統計及び解析計画 | 9 統計学的事項 |
| 9.7.2 | 症例数の決定 | 9.8 参加者数の設定 |
| 9.8 | 治験の実施又は計画された解析に関する変更 | 9.9 試験実施計画書からの逸脱 |
ICH M11:電子的に構造化・調和された臨床試験実施計画書(ドラフト版)

| Structure and Content of Clinical Study Reports — ICH-E3 | Main Reference Sections: Clinical Electronic Structured Harmonised Protocol — ICH-M11 (draft version) | |
| 1 | Title Page | 1 PROTOCOL SUMMARY |
| 2 | Synopsis | 1 PROTOCOL SUMMARY |
| 3 | Table of Contents for the Individual Clinical Study Report | |
| 4 | List of Abbreviations and Definition of Terms | 14 APPENDIX: GLOSSARY OF TERMS |
| 5 | Ethics | 10 GENERAL CONSIDERATIONS: REGULATORY, ETHICAL, AND TRIAL OVERSIGHT |
| 5.1 | Independent Ethics Committee (IEC) or Institutional Review Board (IRB) | 10.2 Committees |
| 5.2 | Ethical Conduct of the Study | 10.1 Regulatory and Ethical Considerations |
| 5.3 | Patient Information and Consent | 10.3 Informed Consent Process |
| 6 | Investigators and Study Administrative Structure | |
| 7 | Introduction | 2 INTRODUCTION |
| 8 | Study Objectives | 3 TRIAL OBJECTIVES, ENDPOINTS AND ESTIMANDS |
| 9 | Investigational Plan | 4 TRIAL DESIGN |
| 9.1 | Overall Study Design and Plan – Description | 4.1 Description of Trial Design |
| 9.2 | Discussion of Study Design, Including the Choice of Control Groups | 4.2 Rationale for Trial Design |
| 9.3 | Selection of Study Population | 5.1 Selection of Trial Population |
| 9.3.1 | Inclusion Criteria | 5.3 Inclusion Criteria |
| 9.3.2 | Exclusion Criteria | 5.4 Exclusion Criteria |
| 9.3.3 | Removal of Patients from Therapy or Assessment | 7 DISCONTINUATION OF TRIAL INTERVENTION AND PARTICIPANT WITHDRAWAL FROM TRIAL |
| 9.4 | Treatments | 6 TRIAL INTERVENTION AND CONCOMITANT THERAPY |
| 9.4.1 | Treatments Administered | 6.1 Description of Trial Intervention |
| 9.4.2 | Identity of Investigational Product(s) | 6.5 Preparation, Handling, Storage and Accountability |
| 9.4.3 | Method of Assigning Patients to treatment Groups | 6.6 Participant Assignment, Randomisation and Blinding |
| 9.4.4 | Selection of Doses in the Study | 6.3 Dosing and Administration |
| 9.4.5 | Selection and Timing of Dose for Each Patient | 6.3.1 Trial Intervention Dose Modification |
| 9.4.6 | Blinding | 6.6.3 Blinding and Unblinding |
| 9.4.7 | Prior and Concomitant Therapy | 6.8 Concomitant Therapy |
| 9.4.8 | Treatment Compliance | 6.7 Trial Intervention Compliance |
| 9.5 | Efficacy and Safety Variables | 8 TRIAL ASSESSMENTS AND PROCEDURES |
| 9.5.1 | Efficacy and Safety Measurements Assessed and Flow Chart | 8.2 Efficacy Assessments and Procedures |
| 9.5.2 | Appropriateness of Measurements | 4.2 Rationale for Trial Design |
| 9.5.3 | Primary Efficacy Variable(s) | 8.2 Efficacy Assessments and Procedures |
| 9.5.4 | Drug Concentration Measurements | 8.7 Pharmacokinetics |
| 9.6 | Data Quality Assurance | 11.2 Data Quality Assurance |
| 9.7 | Statistical Methods Planned in the Protocol and Determination of Sample Size | 9 STATISTICAL CONSIDERATIONS |
| 9.7.1 | Statistical and Analytical Plans | 9 STATISTICAL CONSIDERATIONS |
| 9.7.2 | Determination of Sample Size | 9.8 Sample Size Determination |
| 9.8 | Changes in the Conduct of the Study or Planned Analyses | 9.9 Protocol Deviations |
ICH M11: Clinical Electronic Structured Harmonised Protocol — Draft version

薬審第335号:治験の総括報告書の構成と内容
ICH-E3: Structure and Content of Clinical Study Reports
| 1 | 標題ページ | 1 | Title Page |
| 2 | 概要 | 2 | Synopsis |
| 3 | 目次 | 3 | Table of Contents for the Individual Clinical Study Report |
| 4 | 略号及び用語の定義一覧 | 4 | List of Abbreviations and Definition of Terms |
| 5 | 倫理 | 5 | Ethics |
| 5.1 | 治験審査委員会(IRB) | 5.1 | Independent Ethics Committee (IEC) or Institutional Review Board (IRB) |
| 5.2 | 治験の倫理的実施 | 5.2 | Ethical Conduct of the Study |
| 5.3 | 患者への情報及び同意 | 5.3 | Patient Information and Consent |
| 6 | 治験責任医師等及び治験管理組織 | 6 | Investigators and Study Administrative Structure |
| 7 | 緒言 | 7 | Introduction |
| 8 | 治験の目的 | 8 | Study Objectives |
| 9 | 治験の計画 | 9 | Investigational Plan |
| 9.1 | 治験の全般的デザイン及び計画―記述 | 9.1 | Overall Study Design and Plan – Description |
| 9.2 | 対照群の選択を含む治験デザインについての考察 | 9.2 | Discussion of Study Design, Including the Choice of Control Groups |
| 9.3 | 治験対象集団の選択 | 9.3 | Selection of Study Population |
| 9.3.1 | 組み入れ基準 | 9.3.1 | Inclusion Criteria |
| 9.3.2 | 除外基準 | 9.3.2 | Exclusion Criteria |
| 9.3.3 | 患者の治療又は評価の打ち切り | 9.3.3 | Removal of Patients from Therapy or Assessment |
| 9.4 | 治療法 | 9.4 | Treatments |
| 9.4.1 | 治療法 | 9.4.1 | Treatments Administered |
| 9.4.2 | 治験薬の同定 | 9.4.2 | Identity of Investigational Product(s) |
| 9.4.3 | 治療群への患者の割付け方法 | 9.4.3 | Method of Assigning Patients to treatment Groups |
| 9.4.4 | 治験における用量の選択 | 9.4.4 | Selection of Doses in the Study |
| 9.4.5 | 各患者の用量の選択及び投与時期 | 9.4.5 | Selection and Timing of Dose for Each Patient |
| 9.4.6 | 盲検化 | 9.4.6 | Blinding |
| 9.4.7 | 前治療及び併用療法 | 9.4.7 | Prior and Concomitant Therapy |
| 9.4.8 | 治療方法の遵守 | 9.4.8 | Treatment Compliance |
| 9.5 | 有効性及び安全性の項目 | 9.5 | Efficacy and Safety Variables |
| 9.5.1 | 有効性及び安全性の評価項目及びフローチャート | 9.5.1 | Efficacy and Safety Measurements Assessed and Flow Chart |
| 9.5.2 | 測定項目の適切性 | 9.5.2 | Appropriateness of Measurements |
| 9.5.3 | 有効性の主要評価項目 | 9.5.3 | Primary Efficacy Variable(s) |
| 9.5.4 | 薬物濃度の測定 | 9.5.4 | Drug Concentration Measurements |
| 9.6 | データの品質保証 | 9.6 | Data Quality Assurance |
| 9.7 | 治験実施計画書で計画された統計手法及び症例数の決定 | 9.7 | Statistical Methods Planned in the Protocol and Determination of Sample Size |
| 9.7.1 | 統計及び解析計画 | 9.7.1 | Statistical and Analytical Plans |
| 9.7.2 | 症例数の決定 | 9.7.2 | Determination of Sample Size |
| 9.8 | 治験の実施又は計画された解析に関する変更 | 9.8 | Changes in the Conduct of the Study or Planned Analyses |
| 10 | 治験対象患者 | 10 | Study Patients |
| 10.1 | 患者の内訳 | 10.1 | Disposition of Patients |
| 10.2 | 治験実施計画書からの逸脱 | 10.2 | Protocol Deviation |
| 11 | 有効性の評価 | 11 | Efficacy Evaluation |
| 11.1 | 解析したデータセット | 11.1 | Data Sets Analyzed |
| 11.2 | 人口統計学的及び他の基準値の特性 | 11.2 | Demographic and Other Baseline Characteristics |
| 11.3 | 治療の遵守状況の測定 | 11.3 | Measurements of treatment Compliance |
| 11.4 | 有効性に関する成績及び個別患者データ一覧表 | 11.4 | Efficacy Results and Tabulations of Individual Patient Data |
| 11.4.1 | 有効性の解析 | 11.4.1 | Analysis of Efficacy |
| 11.4.2 | 統計・解析上の論点 | 11.4.2 | Statistical/Analytical Issues |
| 11.4.2.1 | 共変量による調整 | 11.4.2.1 | Adjustments for Covariates |
| 11.4.2.2 | 脱落又は欠測値の取扱い | 11.4.2.2 | Handling of Dropouts or Missing Data |
| 11.4.2.3 | 中間解析及びデータモニタリング | 11.4.2.3 | Interim Analyses and Data Monitoring |
| 11.4.2.4 | 多施設共同治験 | 11.4.2.4 | Multicenter Studies |
| 11.4.2.5 | 多重比較・多重性 | 11.4.2.5 | Multiple Comparison/Multiplicity |
| 11.4.2.6 | 患者の「有効性評価の部分集団」の使用 | 11.4.2.6 | Use of an “Efficacy Subset” of Patients |
| 11.4.2.7 | 同等性を示すことを意図した実対照薬を用いた試験 | 11.4.2.7 | Active-Control Studies Intended to Show Equivalence |
| 11.4.2.8 | 部分集団の検討 | 11.4.2.8 | Examination of Subgroups |
| 11.4.3 | 個別反応データの作表 | 11.4.3 | Tabulation of Individual Response Data |
| 11.4.4 | 薬剤の用量、薬物濃度及びそれらと反応との関係 | 11.4.4 | Drug Dose, Drug Concentration, and Relationships to Response |
| 11.4.5 | 薬物―薬物及び薬物―疾患の相互作用 | 11.4.5 | Drug-Drug and Drug-Disease Interactions |
| 11.4.6 | 患者ごとの表示 | 11.4.6 | By-Patient Display |
| 11.4.7 | 有効性の結論 | 11.4.7 | Efficacy Conclusions |
| 12 | 安全性の評価 | 12 | Safety Evaluation |
| 12.1 | 治験薬が投与された症例数、期間及び用量 | 12.1 | Extent of Exposure |
| 12.2 | 有害事象 | 12.2 | Adverse Events (AEs) |
| 12.2.1 | 有害事象の簡潔な要約 | 12.2.1 | Brief Summary of Adverse Events |
| 12.2.2 | 有害事象の表示 | 12.2.2 | Display of Adverse Events |
| 12.2.3 | 有害事象の分析 | 12.2.3 | Analysis of Adverse Events |
| 12.2.4 | 患者ごとの有害事象の一覧表 | 12.2.4 | Listing of Adverse Events by Patient |
| 12.3 | 死亡、その他の重篤な有害事象及び他の重要な有害事象 | 12.3 | Deaths, Other Serious Adverse Events, and Other Significant Adverse Events |
| 12.3.1 | 死亡、その他の重篤な有害事象及び他の重要な有害事象の一覧表 | 12.3.1 | Listing of Deaths, Other Serious Adverse Events and Other Significant Adverse Events |
| 12.3.1.1 | 死亡 | 12.3.1.1 | Deaths |
| 12.3.1.2 | その他の重篤な有害事象 | 12.3.1.2 | Other Serious Adverse Events |
| 12.3.1.3 | 他の重要な有害事象 | 12.3.1.3 | Other Significant Adverse Events |
| 12.3.2 | 死亡、その他の重篤な有害事象及び他のいくつかの重要な有害事象の叙述 | 12.3.2 | Narratives of Deaths, Other Serious Adverse Events and Certain Other Significant Adverse Events |
| 12.3.3 | 死亡、その他の重篤な有害事象及び他の重要な有害事象の分析及び考察 | 12.3.3 | Analysis and Discussion of Deaths, Other Serious Adverse Events and Other Significant Adverse Events |
| 12.4 | 臨床検査値の評価 | 12.4 | Clinical Laboratory Evaluation |
| 12.4.1 | 患者ごとの個々の臨床検査異常値の一覧表(14.3.4) | 12.4.1 | Listing of Individual Laboratory Measurements by Patient (16.2.8) and Each Abnormal Laboratory Value (14.3.4) |
| 12.4.2 | 各臨床検査項目の評価 | 12.4.2 | Evaluation of Each Laboratory Parameter |
| 12.4.2.1 | 治験期間を通しての臨床検査値 | 12.4.2.1 | Laboratory Values Over Time |
| 12.4.2.2 | 個々の患者の変化 | 12.4.2.2 | Individual Patient Changes |
| 12.4.2.3 | 個々の臨床的に重要な異常 | 12.4.2.3 | Individual Clinically Significant Abnormalities |
| 12.5 | バイタルサイン、身体的所見及び安全性に関連する他の観察項目 | 12.5 | Vital Signs, Physical Findings and Other Observations Related to Safety |
| 12.6 | 安全性の結論 | 12.6 | Safety Conclusions |
| 13 | 考察と全般的結論 | 13 | Discussion and Overall Conclusions |
| 14 | 本文中には含めないが、引用する表、図及びグラフ | 14 | Tables, Figures and Graphs Referred to but Not Included in the Text |
| 14.1 | 人口統計学的データ | 14.1 | Demographic Data |
| 14.2 | 有効性データ | 14.2 | Efficacy Data |
| 14.3 | 安全性データ | 14.3 | Safety Data |
| 14.3.1 | 有害事象の表示 | 14.3.1 | Displays of Adverse Events |
| 14.3.2 | 死亡、その他の重篤な有害事象及び他の重要な有害事象の一覧表 | 14.3.2 | Listings of Deaths, Other Serious and Significant Adverse Events |
| 14.3.3 | 死亡、その他の重篤な有害事象及び他の特に重要な有害事象の叙述 | 14.3.3 | Narratives of Deaths, Other Serious and Certain Other Significant Adverse Events |
| 14.3.4 | 患者ごとの個々の臨床検査異常値の一覧表 | 14.3.4 | Abnormal Laboratory Value Listing (each patient) |
| 15 | 引用文献の一覧表 | 15 | Reference List |
| 16 | 付録 | 16 | Appendices |
| 16.1 | 治験に関する情報 | 16.1 | Study Information |
| 16.1.1 | 治験実施計画書及びその改訂 | 16.1.1 | Protocol and protocol amendments |
| 16.1.2 | 症例記録用紙の見本(内容の異なるページのみ) | 16.1.2 | Sample case report form (unique pages only) |
| 16.1.3 | 治験審査委員会の一覧(確認が行われた年月日、並びに委員の氏名及び職名)、患者への説明文書及び同意書の見本 | 16.1.3 | List of IECs or IRBs (plus the name of the committee Chair if required by the regulatory authority) – Representative written information for patient and sample consent forms |
| 16.1.4 | 治験責任医師及び他の重要な治験参加者の一覧表及び説明(簡潔な(1ページ) 履歴書又は治験の実施に関連する訓練や経験についての履歴書と同等の要約を含む) | 16.1.4 | List and description of investigators and other important participants in the study, including brief (1 page) CVs or equivalent summaries of training and experience relevant to the performance of the clinical study |
| 16.1.5 | 治験総括(調整)医師又は治験依頼者の医学責任者の署名 | 16.1.5 | Signatures of principal or coordinating investigator(s) or sponsor’s responsible medical officer depending on the regulatory authority’s requirement |
| 16.1.6 | 複数のロットが用いられた場合には、治験に用いられたロットごとの薬剤を投与された患者一覧表 | 16.1.6 | Listing of patients receiving test drug(s)/investigational product(s) from specific batches where more than one batch was used |
| 16.1.7 | 無作為化の方法及びコード(患者の識別及び割り付けられた治療) | 16.1.7 | Randomization scheme and codes (patient identification and treatment assigned) |
| 16.1.8 | 監査手順に関する資料、監査証明書(可能であれば) | 16.1.8 | Audit certificates (if available) |
| 16.1.9 | 統計手法に関する文書 | 16.1.9 | Documentation of statistical methods |
| 16.1.10 | 臨床検査に関して施設間の標準化及び品質保証を行ったのであればその方法と手順に関する文書 | 16.1.10 | Documentation of inter-laboratory standardization methods and quality assurance procedures if used |
| 16.1.11 | 治験に基づく公表文献 | 16.1.11 | Publications based on the study |
| 16.1.12 | 総括報告書で引用された重要な公表文献 | 16.1.12 | Important publications referenced in the report |
| 16.2 | 患者データ一覧表 | 16.2 | Patient Data Listings |
| 16.2.1 | 中止症例 | 16.2.1 | Discontinued patients |
| 16.2.2 | 治験実施計画から逸脱した症例 | 16.2.2 | Protocol deviations |
| 16.2.3 | 有効性の解析から除外された症例 | 16.2.3 | Patients excluded from the efficacy analysis |
| 16.2.4 | 人口統計学的データ | 16.2.4 | Demographic data |
| 16.2.5 | 服薬遵守及び(又は)薬物濃度データ(可能であれば) | 16.2.5 | Compliance and/or drug concentration data (if available) |
| 16.2.6 | 個々の有効性反応データ | 16.2.6 | Individual efficacy response data |
| 16.2.7 | 患者ごとの有害事象一覧表 | 16.2.7 | Adverse event listings (each patient) |
| 16.3 | 症例記録 | 16.3 | Case Report Forms |
| 16.3.1 | 死亡、その他の重篤な有害事象発現例及び有害事象による投与中止例の症例記録 | 16.3.1 | CRFs for deaths, other serious adverse events and withdrawals for AE |
| 16.3.2 | 提出された他の症例記録 | 16.3.2 | Other CRFs submitted |





