


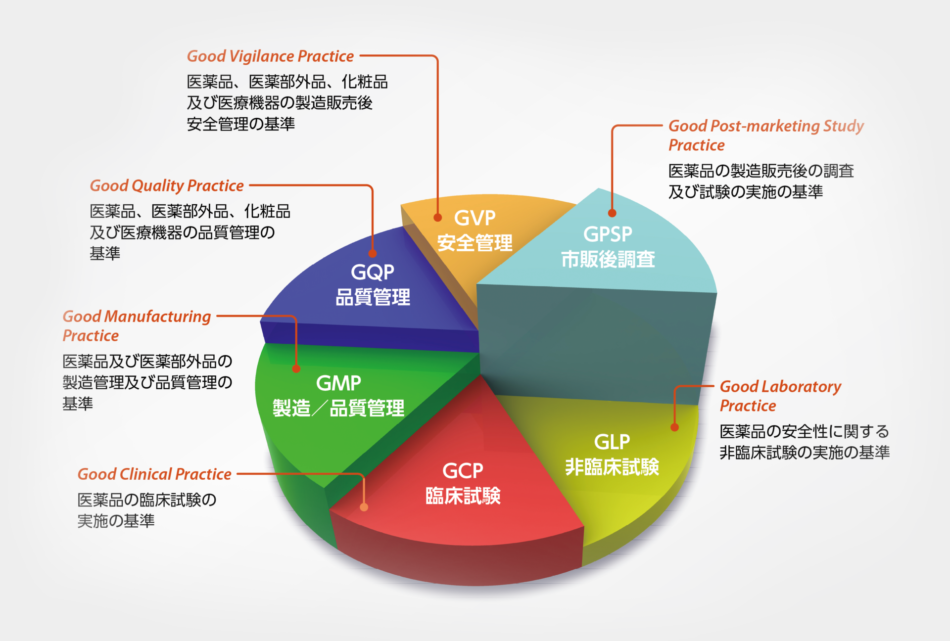
医薬翻訳
当社では、医学・薬学の専門知識と医薬品開発経験を有するグローバルな専門家ネットワークを活用して、非臨床試験から承認申請、そして市販後調査に至る一連の資料の翻訳を行っています。
当社は、ターゲット言語を母国語とする世界中の翻訳者と密接な協力関係を構築しているため、各専門分野の原稿を最適任者に割り当てることができます。翻訳者は、各自の専門分野に絞って翻訳を進めるため、内容や専門用語の調査に時間をかけず、スムーズで効率的な翻訳作業をしています。
翻訳者による翻訳後は、当社QCスタッフが、品質を保証するために、訳文の厳重な品質チェックを行っています。また、当社QCスタッフと翻訳者は、ICH国際医薬用語集(MedDRA)、TRADOS翻訳メモリ、各案件固有の用語集を活用することで、大規模プロジェクトでも個々の翻訳の正確性と一貫性を保っています。
最終翻訳物の納品後は、QC点検証跡、品質管理証明書、翻訳証明書を提出することも可能です。

Medical and Pharmaceutical Translation
MedicaLingual utilizes a global network of professionals with a high level of medical and pharmaceutical knowledge and an experience in the field of drug development to provide translation service for documents across the entire drug development process, from non-clinical study to new drug application and post-marketing surveillance.
Close cooperation between MedicaLingual and native translators in the world provides a full range of experienced expertise, making it possible to assign translations in each specialized field to the most qualified professional. Translators focus on specialized areas to ensure smooth and efficient translation that reduces the time required to understand content and verify specialized terms.
After translation, our QC staff conducts strict checks to ensure the final product meets the highest standards. In addition, our QC staff and individual translators utilize the Medical Dictionary for Regulatory Activities Terminology (MedDRA), TRADOS translation memories and specific terms for each project to ensure that each translation is both accurate and consistent across even the largest projects.
MedicaLingual can present the client with QC inspection traceability assurance, a quality control certificate or a translation certificate for each final deliverable.
主な翻訳文書の入門的な解説
CMC(化学・製造・品質管理)
製造方法並びに規格及び試験方法
これらの試験は、医薬品がどのような物質であり、どのような性質を有しているかを明らかにするために行われるものであり、言うなれば当該医薬品の品質上の「身分証明書」に当たるものです。
具体的には、構造決定、物理化学的特性の確認、製造方法、どのような規格に基づいて製造されているか、さらにそれらがどのような試験によって確認されるか、といった事項が対象となります。
構造決定は、物質を構成する原子の配置を明らかにすることにより、その薬の化学構造を特定するものです。
このための方法としては、NMR(核磁気共鳴法)やX線結晶構造解析などが用いられます。
また、物理化学的特性の確認には、各医薬品の特性に応じた適切な試験法が用いられます。これらの試験法は、日本薬局方などに定められています。
規格には、薬の有効成分そのものである原薬の規格と、これに賦形剤や添加物などを加えて製品化した製剤の規格があります。
これらの規格では、例えば以下のような項目が設定されます。
名称、構造式又は示性式、分子式及び分子量、基原、含量規格、性状、確認試験、示性値(物理的・化学的性質を含む)、純度試験、水分又は乾燥減量、強熱残分、灰分又は酸不溶性灰分、製剤試験、特殊試験、その他の試験項目(例えば微生物限度試験、原薬の粒子径)、定量法、標準物質、試薬、試液です。
なお、構造決定や物理化学的特性に関する情報は、その後、当該医薬品の添付文書にも記載されます。
また、規格については、「新医薬品の規格及び試験方法の設定について」というガイドラインが示されており、これはICHの英語版ガイダンスにも対応しています。
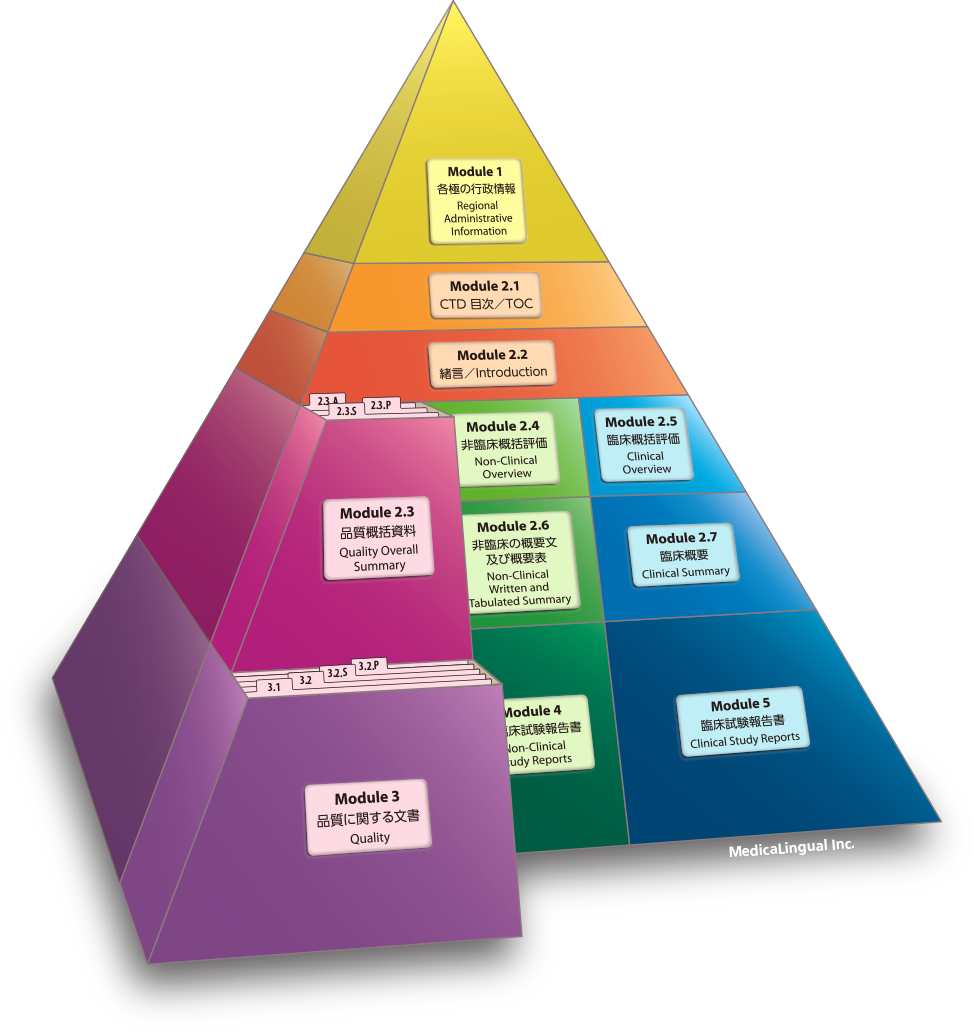
An Introductory Overview of Major Translation Document Types
Chemistry, Manufacturing, and Controls (CMC)
Manufacturing methods, specifications and testing methods
These studies serve, so to speak, as the “identification record” of a drug substance, clarifying what kind of entity the drug is and what characteristics it has as a substance. They cover such matters as structure determination, confirmation of physicochemical properties, the manufacturing method, the specifications according to which the product is manufactured, and the tests used to demonstrate compliance with those specifications.
Structure determination establishes the chemical structure of the drug by elucidating the arrangement of atoms within the substance. Methods used for this purpose include NMR (nuclear magnetic resonance spectroscopy) and X-ray crystallography. Confirmation of physicochemical properties is performed using test methods appropriate to the characteristics of each drug. These test methods are specified in sources such as the Japanese Pharmacopoeia.
Specifications include those for the drug substance, which is the active ingredient of the drug, and those for the drug product, which is the finished product prepared by adding excipients, additives, and other components to the drug substance. Specifications are established for the following types of items:
name; structural formula or rational formula; molecular formula and molecular weight; origin; content specification; properties; identification test; specific physical and chemical value (including physicochemical properties); purity test; water content or loss on drying; residue on ignition, ash, or acid-insoluble ash; pharmaceutical preparation test; specific test; other test items (such as microbial limit test and particle size of the drug substance); assay; reference material; and agent / test solution.
Information on structure determination and physicochemical properties is later included in the package insert for the drug.
With respect to specifications, there is a guideline entitled “Setting of Specifications and Test Methods for New Drug Substances and New Drug Products,” which corresponds to the English ICH guidance.

安定性試験
安定性試験は、医薬品を一定の条件下で一定期間保存した際に、品質がどのように変化するかを評価するために実施される試験です。これらの試験で得られたデータに基づき、製品の貯蔵条件や有効期間が設定されます。
安定性試験には、原薬を対象とするものと、製剤を対象とするものがあります。主な試験として、以下のものが挙げられます。
長期保存試験:申請される、又は承認されたリテスト期間若しくは有効期間を設定するために、表示ラベルに記載される貯蔵条件の下で実施される安定性試験です。
苛酷試験:原薬の本質的な安定性を明らかにするために実施される試験です。通常、開発段階で行われ、加速試験よりも厳しい保存条件を用いて実施されます。
また、製剤については、苛酷条件による影響を評価するために実施され、光安定性試験や、特定の剤形(例えば計量吸入剤、クリーム剤など)に応じた特殊な試験が含まれます。
加速試験:原薬又は製剤における化学的変化又は物理化学的変化を促進する保存条件を用いて実施される試験です。加速試験の結果は、長期保存試験の結果とあわせて、申請された貯蔵条件で長期間保存した場合に想定される化学的影響を評価するために用いられます。
なお、安定性試験については、「安定性試験ガイドラインの改訂について」というガイドラインが示されており、これはICHの英語版ガイダンスにも対応しています。
Stability tests
A stability test is a study conducted to examine how a drug changes when stored under various conditions and for various periods of time. Based on the data obtained from these studies, the storage conditions and shelf life of the finished product are established. These studies include those relating to the drug substance and those relating to the drug product. Stability tests include the following types of studies:
Long-term shelf-life testing: A stability study conducted under the storage conditions indicated on the label in order to establish the proposed or approved retest period or shelf life.
Stress testing: A study performed to elucidate the intrinsic stability of the drug substance. It is conducted during the development stage and is normally carried out under storage conditions more severe than those used for accelerated testing.
For the drug product, it is also performed to assess the effects of severe conditions and includes photostability testing and specific tests for particular dosage forms, such as metered-dose inhalers and creams.
Accelerated testing: A study conducted using storage conditions that accelerate the chemical change or physicochemical change of the drug substance or drug product. The results of accelerated testing, together with those of long-term shelf-life testing, are used to evaluate the chemical effects of long-term storage under the proposed storage conditions.
With respect to stability tests, there is a guideline entitled “Revision of the Guideline for Stability Testing,” which corresponds to the English ICH guidance.

非臨床試験
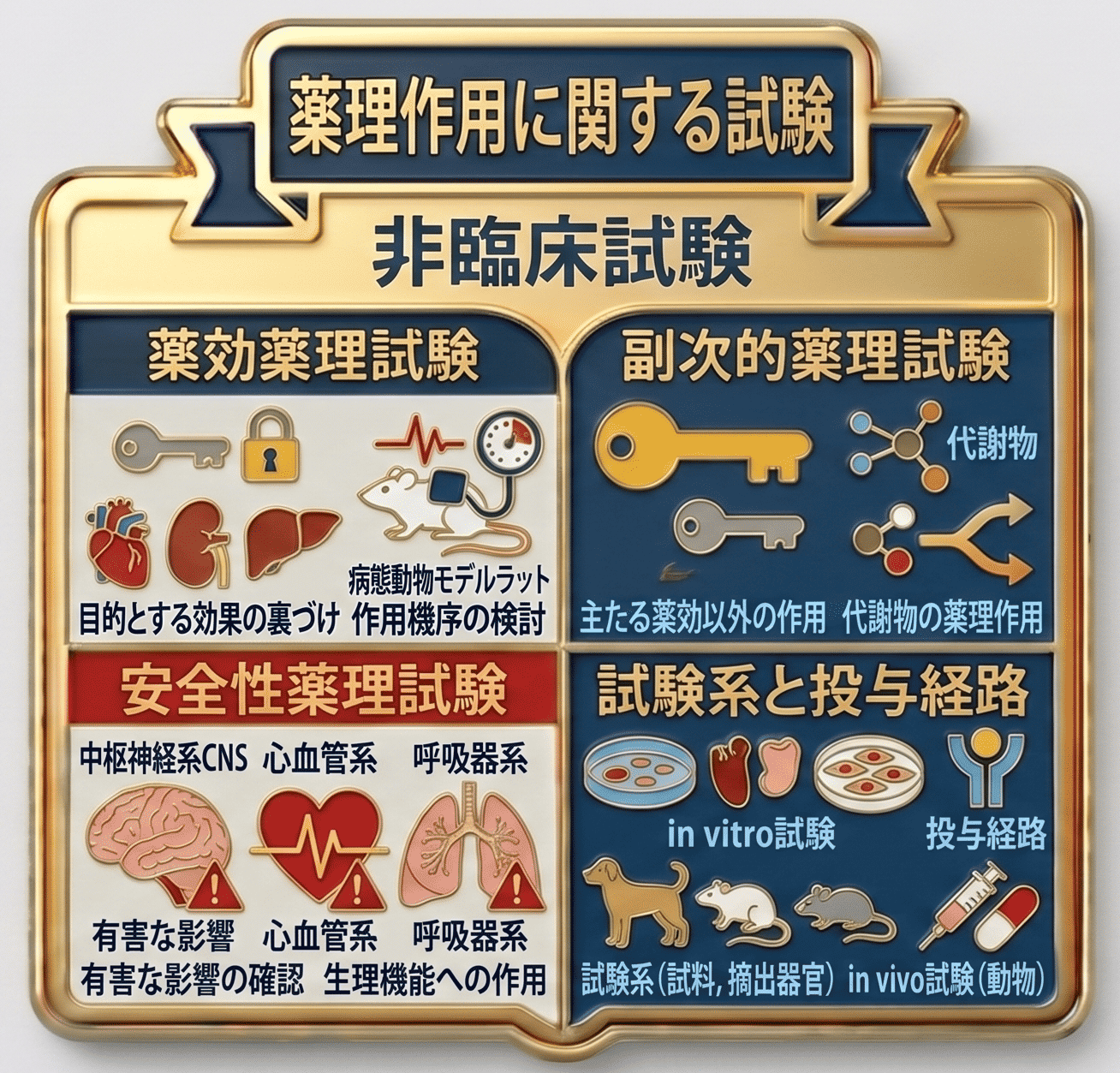
薬理作用に関する試験
これらの試験は、医薬品が示す薬理作用を評価するために実施されるものであり、大きく以下の3つに分類されます。
薬効薬理試験
薬理作用とは、まず当該医薬品の存在意義となる主たる薬効を意味します。薬効薬理試験は、その医薬品が目的とする効果を裏づけるために実施される試験です。
本試験では、薬理作用が体内でどのように発現するかという作用機序、どの用量で、どの投与経路により、どの動物種に投与した場合に、どの程度の効果が認められるかなどを検討します。
通常、これらの評価は、まずin vitroで臓器片や組織などを用いて行われ、その後、ラット、マウス、ネコ、イヌなどの動物を用いたin vivo試験で検討されます。さらに、健康な動物における評価に加え、ヒトの病態を想定して作製された病態動物モデル(例えば高血圧ラット、関節炎モデルなど)を用いた検討も行われます。
薬理作用は医薬品ごとに固有であるため、その評価に用いられる試験方法も各医薬品の特性に応じて異なります。
副次的薬理試験
物質は、目的とする薬理作用に加えて、副次的な作用を有する場合があるため、そのような作用を評価するのが副次的薬理試験です。
また、医薬品が体内に取り込まれた後、代謝によって生じる代謝物が薬理作用を示す場合もあり、これについてもあわせて検討されます。
安全性薬理試験
一方で、期待される薬効が得られたとしても、ヒトに適用した際に他の臓器又は生理機能に有害な影響を及ぼす場合には、医薬品として望ましいものではありません。こうした望ましくない作用がないことを確認するために実施されるのが安全性薬理試験です。
安全性薬理試験では、治療用量及びそれを上回る用量において、被験物質が生理機能に及ぼす望ましくない薬力学的作用を評価します。
試験には、動物モデル、又は動物若しくはヒト由来の試料(摘出器官及び組織、培養細胞、細胞フラグメント、受容体など)を用いたin vitro系が用いられます。また、in vivo試験では、臨床で想定される投与経路により被験物質が投与されます。
主な評価項目としては、中枢神経系(CNS)、心血管系、呼吸器系などに対する影響が挙げられます。
Non-clinical Study
Studies on pharmacological effects
These studies are conducted to evaluate the pharmacological effects exhibited by a medicinal product and are broadly classified into the following three categories.
Primary pharmacodynamic studies
Pharmacological effects primarily refer to the principal efficacy that constitutes the rationale for the existence of the medicinal product. Primary pharmacodynamic studies are conducted to substantiate the intended effect of the medicinal product.
These studies examine such aspects as the mechanism of action by which the pharmacological effect is expressed in the body, the dose at which the effect is observed, the route of administration, the animal species to which the medicinal product is administered, and the extent of the effect observed.
In general, these evaluations are first conducted in vitro using tissue or organ preparations and are then investigated in in vivo studies using animals such as rats, mice, cats, and dogs. In addition to evaluations in healthy animals, studies are also conducted using disease animal models established to simulate human pathological conditions (for example, hypertensive rats and arthritis models).
Because pharmacological effects are specific to each medicinal product, the test methods used for their evaluation also differ depending on the characteristics of each product.
Secondary pharmacodynamic studies
Because a substance may have not only the intended pharmacological effect but also secondary effects, secondary pharmacodynamic studies are conducted to evaluate such effects.
In addition, metabolites formed after the medicinal product is taken up into the body may also exhibit pharmacological effects, and these are likewise evaluated.
Safety pharmacology studies
On the other hand, even if the expected efficacy is obtained, a substance is not desirable as a medicinal product if it causes harmful effects on other organs or physiological functions when administered to humans. Safety pharmacology studies are conducted to confirm the absence of such undesirable effects.
In safety pharmacology studies, the undesirable pharmacodynamic effects of the test substance on physiological functions are evaluated at the therapeutic dose and at doses above the therapeutic dose.
These studies use animal models or in vitro systems employing animal- or human-derived materials (including isolated organs and tissues, cultured cells, cell fragments, and receptors). In in vivo studies, the test substance is administered by the route intended for clinical use.
The principal evaluation parameters include effects on the central nervous system (CNS), cardiovascular system, and respiratory system.
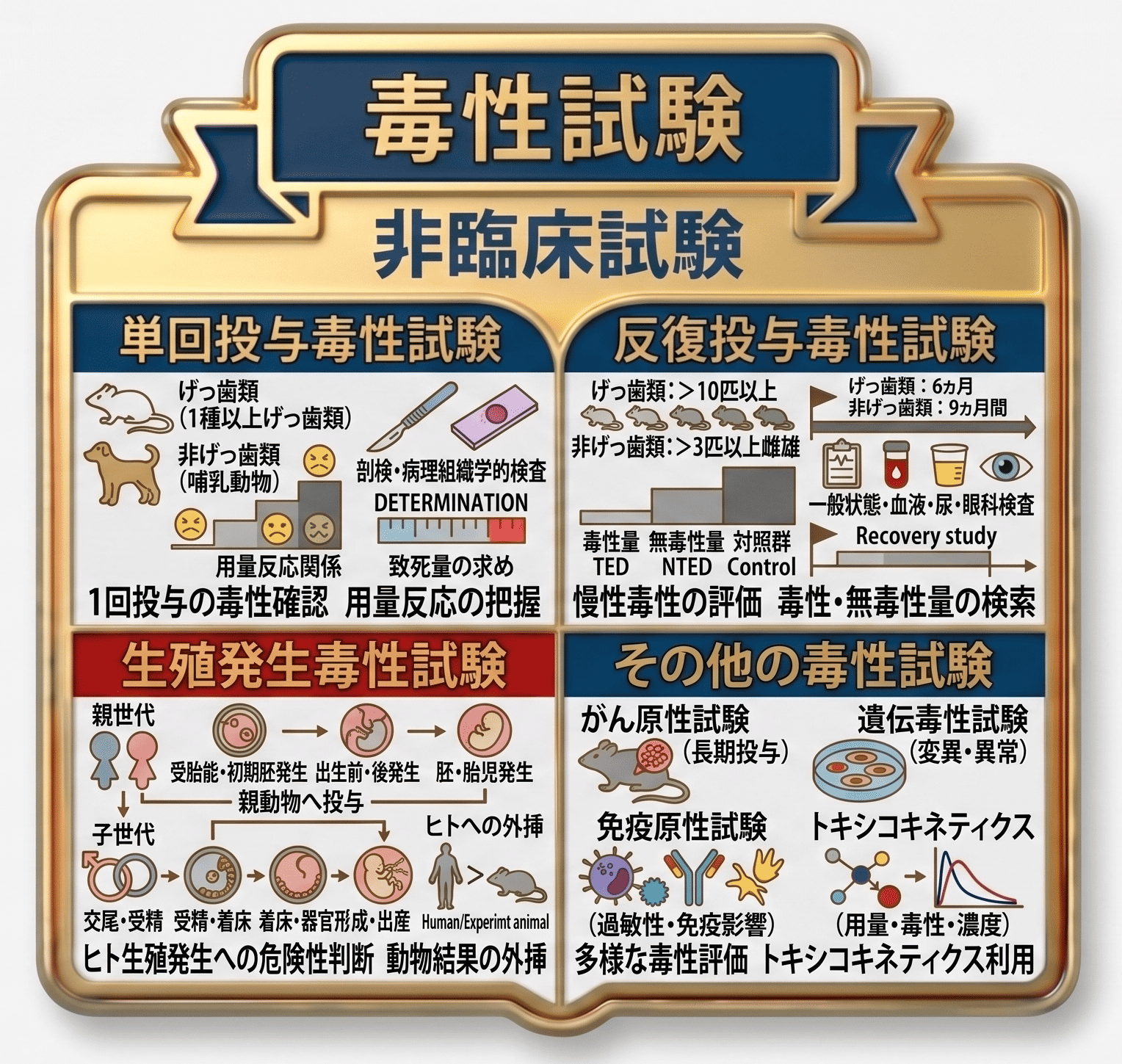
毒性試験
毒性試験は、被験物質を動物に投与し、有害な作用、すなわち毒性の有無及びその内容を評価することにより、医薬品としてヒトに投与した際の安全性を確保するために実施される試験です。主な試験として、単回投与毒性試験、反復投与毒性試験、生殖発生毒性試験があり、このほか、がん原性試験、遺伝毒性試験、免疫原性試験、トキシコキネティクス試験などがあります。
多くの毒性試験については、ICHにおいて国際的な整合化が進められており、国内のガイドラインもおおむねICHガイドラインに対応しています。
1. 単回投与毒性試験
この試験は、被験物質を哺乳類に1回のみ投与した際に生じる毒性、すなわち単回投与毒性(急性毒性ともいう)を明らかにすることを目的としています。
試験には2種以上の動物種が用いられ、主としてマウス及びラットが選択されます。少なくとも1種については雌雄を含め、原則としてヒトで予定される投与経路により投与します。
用量段階は、毒性の徴候が把握でき、かつ用量反応関係が認められるように設定されます。すなわち、用量の増加に伴って毒性徴候が強まる関係が確認できるように設計されます。
試験期間中(通常14日間)は、毒性徴候の種類、程度、推移及び可逆性について、用量との関連を踏まえて観察・記録します。観察期間中に死亡した動物及び試験終了時の全生存げっ歯類については、剖検を行います。剖検時に肉眼的異常が認められた場合には、病理組織学的検査を実施します。
本試験の結果から、被験物質のおおよその致死量を求め、ヒトにおける毒性予測の参考情報とします。
2. 反復投与毒性試験
反復投与毒性試験(慢性毒性試験ともいう)は、被験物質を哺乳類に反復して投与した後に認められる毒性変化(徴候等)を記録するとともに、毒性変化を生じる用量(毒性量)及び毒性変化を生じない用量(無毒性量)を検索することを目的としています。
この試験では2種以上の動物を用い、げっ歯類では1群当たり雌雄各10匹以上、非げっ歯類では雌雄各3匹以上を基本とします。投与は、ヒトで予定される投与経路により、想定される使用期間に応じて、げっ歯類では6か月間、非げっ歯類では9か月間実施されます。
用量段階は少なくとも3段階設定され、毒性量及び無毒性量の双方を含み、かつ用量反応関係が認められるように設定されます。また、比較対照として、被験物質を投与しない対照群を設け、溶媒又はmediumのみを投与します。
主な観察及び検査項目としては、一般状態(clinical symptoms)、体重、摂餌量、飲水量、血液検査、尿検査、眼科的検査、その他の機能検査などが挙げられます。なお、毒性変化の可逆性を確認するため、必要に応じて回復性試験が実施されることがあります。
投与期間中に死亡した動物及び著しく衰弱したdebilitated animalについては、その時点で剖検を行います。さらに、投与終了時には全例について剖検を実施し、あわせて病理組織学的検査を行います。
3. 生殖発生毒性試験
この試験は、被験物質が哺乳類の生殖及び発生に及ぼす影響を評価し、その結果を他の毒性試験及び薬理試験の成績とあわせて検討することにより、ヒトの生殖発生に対するリスク評価のための情報を提供することを目的としています。
親動物にはさまざまな時期に被験物質を投与し、親世代の生殖機能及び子世代への影響を検討します。投与期間に応じて、これらの試験は3種類に分けられます。すなわち、着床までを対象とする「受胎能及び着床までの初期胚発生に関する試験」、出生前及び出生後の発生並びに母体機能を評価する試験、並びに器官形成期終了までを対象とする「胚・胎児発生に関する試験」です。
試験の種類に応じて、以下のような項目が観察されます。
A. 交尾前~受精
[親世代の生殖機能、配偶子の発生及び成熟、交尾行動、受精]
B. 受精~着床
[親の生殖機能、着床前発生、着床]
C. 着床~硬口蓋閉鎖
[親の生殖機能、胚発生、主要器官の形成]
D. 硬口蓋閉鎖~妊娠終了
[親の生殖機能、胎児発生と成長、器官発生を含む器官の発生・成長]
E. 出生~離乳
[親の生殖機能、新生児の子宮外生活への適応、離乳前の発生と成長]
F. 離乳~性成熟
[離乳後の発生と成長、自立生存への適応、完全な性機能の獲得]
ヒトと実験動物では、妊娠期間や薬物代謝が異なるほか、動物種ごとに特徴的な奇形も存在するため、動物で得られた結果をそのままヒトに当てはめることはできません。したがって、ヒトの生殖に対する影響をより適切に検出できるよう、投与経路、用量、投与期間、観察項目等を考慮して動物試験が設計されます。
また、動物で影響が認められた用量がヒトではどの程度に相当するかを推定することを、動物試験成績をヒトに外挿するといいます。
4. その他の毒性試験
上記のほか、がん原性試験、遺伝毒性試験、免疫原性試験などがあります。がん原性試験では、動物に長期間投与することにより、ヒトにおける発がん性の可能性を検討します。遺伝毒性試験では、培養細胞などに被験物質を添加し、細胞の変異の有無を評価します。免疫原性試験では、免疫機能に対する影響を評価し、過敏性やアレルギーなども検討対象となります。
これらの試験が必要となるかどうかは、被験物質の投与期間や薬物の特性に応じて判断され、実施が求められる場合と、必ずしも必要でない場合があります。
さらに、トキシコキネティクス試験では、各種毒性試験における用量と毒性との関係を薬物濃度との関連で検討し、ヒトにおける安全性評価の参考情報とします。
Toxicity studies
Toxicity studies are studies conducted to ensure the safety of a medicinal product when administered to humans by administering a test substance to animals and evaluating the presence and nature of harmful effects, that is, toxicity. The principal studies include single-dose toxicity study, repeated-dose toxicity study, and reproduction toxicity study. In addition, carcinogenicity studies, genotoxicity studies, immunogenicity studies, and toxicokinetic studies are also conducted.
For many toxicity studies, international harmonization has progressed within ICH, and domestic guidelines generally correspond to the ICH guidelines.
1. Single-dose toxicity study
The purpose of this study is to clarify the toxicity produced when a test substance is administered only once to mammals, that is, single dose toxicity (also referred to as acute toxicity).
Two or more animal species are used in this study, principally mice and rats. At least one species includes both males and females, and, in principle, the test substance is administered by the administration route intended for use in humans.
The dose levels are selected so that toxic signs can be identified and a dose-response relationship can be observed. In other words, the study is designed so that a relationship can be confirmed in which the severity of toxic signs increases as the dose increases.
During the study period (usually 14 days), the type, severity, course, and reversibility of toxic signs are observed and recorded in relation to dose. Animals that die during the observation period and all surviving rodents at the end of the study are subjected to autopsy. If gross abnormalities are observed at autopsy, histopathological examination is performed.
Based on the results of this study, the approximate lethal dose of the test substance is determined and used as reference information for predicting toxicity in humans.
2. Repeated-dose toxicity study
The purpose of the repeated dose toxicity study (also referred to as a chronic toxicity study) is to record toxic changes (including signs) observed after repeated administration of a test substance to mammals and to identify the dose that produces toxic changes (the toxic effect dose) and the dose that does not produce such changes (the non-toxic effect dose).
This study uses two or more animal species, generally with at least 10 males and 10 females per group in rodents and at least 3 males and 3 females per group in non-rodents. Administration is carried out by the route intended for use in humans for 6 months in rodents and 9 months in non-rodents, according to the anticipated duration of clinical use.
At least three dose levels are established, including both a toxic effect dose and a non-toxic effect dose, and are selected so that a dose-response relationship can be observed. In addition, a control group is included for comparison, in which no test substance is administered and only the vehicle or medium is given.
The principal observation and examination items include general condition (clinical symptoms), body weight, feed consumption, water consumption, hematology, urinalysis, ophthalmology, and other functional tests. In addition, a recovery study may be conducted, as necessary, to confirm the reversibility of toxic changes.
Animals that die during the dosing period and any markedly weakened debilitated animal are subjected to autopsy at that time point. Furthermore, at the end of the dosing period, all animals undergo autopsy, and histopathological examination is also performed.
3. Reproduction toxicity study
The purpose of this study is to evaluate the effects of a test substance on reproduction and development in mammals and, by considering the results together with findings from other toxicity studies and pharmacological studies, to provide information for the assessment of risk to human reproduction and development.
The test substance is administered to parent animals at various stages, and the reproductive function of the parental generation and the effects on the offspring generation are evaluated. Depending on the dosing period, these studies are divided into three types: a “study of fertility and early embryonic development to implantation,” a study evaluating prenatal and postnatal development as well as maternal function, and an “embryo-fetal development study,” in which dosing continues until the end of organogenesis.
Depending on the type of study, the following items are observed:
A. Premating to conception
[parental reproductive function, development and maturation of gametes, mating behavior, fertilization]
B. Conception to implantation
[parental reproductive function, preimplantation development, implantation]
C. Implantation to closure of the hard palate
[parental reproductive function, embryonic development, formation of major organs]
D. Closure of the hard palate to the end of pregnancy
[parental reproductive function, fetal development and growth, organ development including organogenesis]
E. Birth to weaning
[parental reproductive function, adaptation of neonates to extrauterine life, preweaning development and growth]
F. Weaning to sexual maturity
[postweaning development and growth, adaptation to independent survival, acquisition of full sexual function]
Because humans and laboratory animals differ in gestation period and drug metabolism, and because characteristic malformations also vary among animal species, results obtained in animals cannot be directly applied to humans. Accordingly, animal studies are designed by taking into account such factors as the route of administration, dose, dosing period, and observation items so that effects on human reproduction can be detected more appropriately.
In addition, estimating the extent to which the dose associated with findings in animals corresponds to that in humans is referred to as extrapolate animal study results to humans.
4. Other toxicity studies
In addition to the above, there are carcinogenicity studies, genotoxicity studies, and immunogenicity studies. Carcinogenicity studies examine the potential for carcinogenicity in humans by long-term administration to animals. Genotoxicity studies evaluate the presence or absence of cellular mutations by adding the test substance to cultured cells and similar systems. Immunogenicity studies assess effects on immune function, including hypersensitivity and allergy.
Whether these studies are required is determined according to the duration of administration of the test substance and the characteristics of the drug; in some cases, their conduct is required, whereas in others it is not necessarily required.
Furthermore, toxicokinetic studies examine the relationship between dose and toxicity in various toxicity studies in relation to drug concentration and provide reference information for the evaluation of safety in humans.
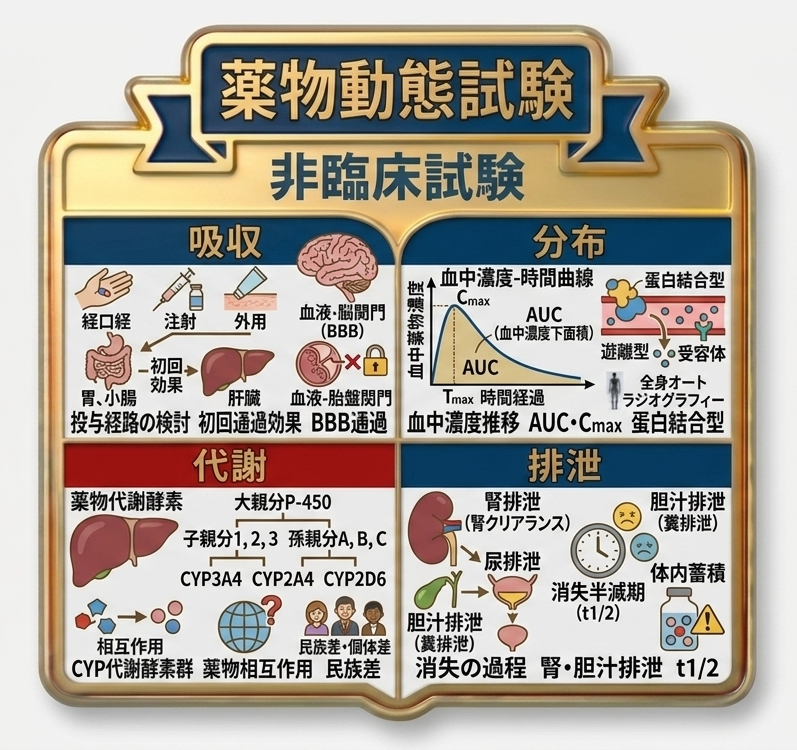
薬物動態試験
薬物動態試験は、薬物が体内に投与されてから作用を示し、最終的に体外へ排出されるまでの過程を評価する試験です。薬物は、投与後に血流によって作用部位へ運ばれ、その部位の受容体と結合して初めて薬効を発現します。したがって、各種投与経路により投与された薬物が、投与用量のうちどの程度、どの経路を経て、どの程度の時間で作用部位に到達するか、また、未変化体のまま作用するのか、あるいは代謝を受けた形で作用するのか、さらに、その後どのように代謝され、どのように排出されるのかを把握することは、有効性及び安全性の評価において重要です。
非臨床薬物動態試験では、主として動物を用いて被験物質の体内動態を明らかにし、その成績をもとにヒトにおける体内動態を予測します。薬物の体内動態は、吸収、分布、代謝及び排泄の4つの過程に区分され、これらを総称してADMEと呼びます。なお、代謝及び排泄は、あわせて消失の過程と整理されます。
1. 吸収は、薬物が投与部位から血液中へ取り込まれる過程です。多くの薬物は経口投与され、胃で溶解した後、小腸で吸収されて血流に入り、作用部位へ運ばれます。ただし、胃酸により分解される薬物もあるため、その場合には腸溶錠などの剤形上の工夫がなされます。また、小腸から吸収された薬物は門脈を経て肝臓に至り、全身循環へ移行する前に代謝されて活性を失う場合があり、これを初回通過効果といいます。これを回避する目的で、口腔粘膜から直接循環血中へ吸収させる舌下錠が用いられることもあります。注射剤は経口投与よりも速やかに作用し、特に静脈内投与では薬物が直接循環血中に入るため、速やかに作用部位へ到達します。筋肉内投与及び皮下投与も、経口投与と比較して速やかな作用発現が期待されます。さらに、血液‐脳関門や血液‐胎盤関門のような生体の防御機構により、薬物の脳や胎盤への移行は制限されるため、これらの部位を標的とする薬物では、移行性を考慮した設計が必要です。
2. 分布は、吸収された薬物が血流により全身へ運ばれ、各組織へ移行する過程です。薬物は標的となる受容体だけでなく、血中蛋白とも結合しますが、蛋白結合した薬物は血管壁を通過しにくく、作用部位へ移行しにくいため、実際に薬効発現に関与するのは蛋白と結合していない遊離型薬物です。血中薬物濃度は投与後に上昇し、最高血中濃度に達した後、時間の経過とともに低下します。この推移を示すものが血中濃度‐時間曲線であり、その下面積であるAUCは、全身循環に移行した薬物量の指標となります。静脈内投与時と経口投与時のAUCを比較することにより、生物学的利用率を評価することができます。また、動物では、放射標識薬物を用いた全身オートラジオグラフィーにより、経時的な組織分布を視覚的に評価することが可能です。
3. 消失は、薬物が体内から除去される過程であり、代謝及び排泄から構成されます。血中薬物濃度が半減するまでの時間は半減期と呼ばれ、薬物の体内滞留時間を示す重要な指標です。水溶性薬物は主として腎臓から尿中へ排泄されますが、腎機能が低下すると薬物が体内に蓄積し、副作用発現のリスクが高くなるため、臨床では用量調整が必要となります。なお、血漿中から尿中へ薬物がどの程度排泄されるかを示す指標が腎クリアランスです。これに対し、脂溶性薬物はそのままでは排泄されにくいため、肝臓で代謝されて極性の高い形に変換された後、代謝物として胆汁又は尿中へ排泄されます。
肝代謝において重要な役割を担うのが薬物代謝酵素であり、特にチトクロムP-450(CYP)系酵素は、多くの薬物の代謝に関与しています。同一の代謝酵素で代謝される複数の薬物を併用すると、相互に代謝を阻害し、血中濃度の上昇を介して副作用の原因となることがあり、これを薬物相互作用といいます。また、薬物代謝酵素には遺伝多型が存在するため、個人ごとに代謝能が異なります。さらに、ヒトでは個体差に加えて民族差も認められるため、欧米等の海外データがそのまま日本人に適用できない場合があります。ICHでは国際的な整合化が進められていますが、こうした民族差をどのように評価し取り扱うかは、国際共同開発における重要な課題です。
Pharmacokinetic studies
Pharmacokinetic studies are studies that investigate the course of a drug from the time it enters the body, exerts its effects, and is ultimately eliminated from the body. After administration, a drug is transported by the bloodstream to its site of action, where it produces its effect only after the drug molecules bind to and interact with the receptor at that site. Therefore, in order to understand both the efficacy and safety of a drug, it is important to determine, for a drug administered by various administration routes, how much of the administered dose reaches the site of action, by what route, and over what period of time; whether the drug exerts its effect as the unchanged drug or in a chemically modified form; and how, after fulfilling its role, it is broken down, that is, metabolized, and then eliminated from the body. In this sense, pharmacokinetic studies can be regarded as a kind of “travel diary” describing the movement and fate of a drug in the body. In nonclinical pharmacokinetic studies, the ADME profile of a test substance is clarified mainly in animals, and the findings are then used to predict its pharmacokinetics in humans. The movement of a drug through the body is divided into four phases: absorption, distribution, metabolism, and excretion. These are collectively referred to as ADME. Metabolism and excretion together constitute the process of elimination.
1. The absorption phase is the process by which a drug is taken up from the site of administration into the bloodstream. Many drugs are administered orally, dissolve in the stomach, are absorbed in the small intestine, enter the circulating blood, and are then transported to the site of action. However, some drugs are degraded by gastric acid, and for such drugs, formulations such as enteric-coated tablets, which dissolve only after reaching the intestine, are used. In addition, drugs absorbed from the small intestine may pass through the portal vein to the liver and be metabolized before entering the systemic circulation, thereby losing their activity. This is known as the first-pass effect. To avoid this effect, sublingual tablets, which are absorbed through the oral mucosa directly into the circulation, may be used. Injectable preparations act more rapidly than orally administered drugs. In particular, following intravenous injection (i.v.), the drug enters the circulation directly and therefore reaches the site of action very quickly. Intramuscular injection (i.m.) and subcutaneous injection (s.c.) also produce effects more rapidly than oral administration. In addition, mechanisms such as the blood-brain barrier (BBB) and the blood-placental barrier restrict drugs from freely entering the brain or placenta, and drugs intended to act at these sites must therefore be designed with such permeability in mind.
2. The distribution phase refers to the process by which an absorbed drug is carried through the body by the bloodstream and reaches the required tissues. A drug binds not only to its target receptor but also to blood proteins such as albumin. Once bound to proteins, the drug cannot readily pass through the vascular wall and is therefore less able to reach the site of action. For this reason, it is the concentration of the unbound, or free, drug that is most important for pharmacological activity. After administration, the blood concentration of a drug rises as absorption proceeds, reaches a maximum concentration, and then declines over time. This change is represented by the blood concentration-time curve. The area under curve (AUC) is an indicator of the amount of drug that has entered the systemic circulation. By comparing the AUC after intravenous administration with that after oral administration, bioavailability can be assessed. In animals, tissue distribution over time can also be examined visually by whole-body autoradiography using a radio-labeled drug.
3. The elimination phase consists of metabolism and excretion, through which the drug is removed from the body. The time required for the blood drug concentration to decrease by half is called the half-life (t1/2), which serves as an indicator of how long the drug remains in the body. Water-soluble drugs are mainly excreted into the urine via the kidneys, but if renal function is impaired, the drug may accumulate in the body, increasing the risk of adverse effects. In such cases, dose adjustment is required. A related index is renal clearance, which indicates the extent to which a drug is excreted from plasma into the urine. Fat-soluble drugs, on the other hand, are not readily excreted as they are. They are therefore metabolized in the liver into more polar and water-soluble forms and then excreted either in bile and feces or in urine as a metabolite.
An important factor in hepatic drug metabolism is the drug metabolizing enzyme system. Although there are some differences between animals and humans, the most important enzymes are those of the cytochrome P-450 family, which play a central role in intracellular oxidation-reduction reactions. When two or more drugs that are metabolized by the same enzyme are administered together, they may inhibit each other’s metabolism. This phenomenon is called a drug interaction and may lead to adverse effects because blood drug concentrations increase. Drug-metabolizing enzymes also show individual variability, and in cases of polymorphism, where a particular enzyme is absent or reduced, drugs metabolized by that enzyme may not be adequately metabolized and may accumulate in the body. In humans, not only individual differences but also ethnic differences exist. As a result, data obtained in other regions, such as Europe or the United States, may not always be directly applicable to Japanese populations. Although ICH has promoted international harmonization among Japan, the United States, and Europe through data sharing and global study programs, how to address such ethnic differences remains an important issue in international drug development.
臨床試験
治験薬概要書
治験薬概要書は、治験責任医師、治験分担医師、治験ナース、治験コーディネーター(CRC)など、治験に関わる医療従事者に向けて作成される、治験薬に関する重要情報の集約資料です。これまでに得られている臨床及び非臨床データを整理してまとめたものであり、被験者の安全を確保しながら、治験を適切に実施するための基礎資料として用いられます。
わかりやすく言えば、治験薬概要書は「この薬はどのような薬で、これまでにどのような試験が行われ、どのような効果や危険性が分かっているのか」を、治験を担当する医療従事者が理解するための手引書です。治験薬はまだ承認前であり、通常診療で使われている薬とは異なるため、担当する医師やスタッフは、その性質や注意点を十分に理解しておく必要があります。その中心的な情報源が治験薬概要書です。
日本の「医薬品の臨床試験の実施の基準」(GCP)では、治験依頼者に対し、規定の試験により得られた資料並びに被験薬の品質、有効性及び安全性に関する情報に基づいて、治験薬概要書を作成することを求めています。少なくとも、被験薬の化学名又は識別記号、品質、毒性、薬理作用その他の被験薬に関する事項、さらに臨床試験が実施されている場合にはその試験成績を記載する必要があります。実際にはこれらに加えて、用法・用量の考え方、既知の副作用、注意すべき検査値の変化、併用薬に関する注意、前臨床試験から示唆されるリスクなど、被験者管理に有用な情報が体系的に記載されます。
治験依頼者は、治験を医療機関に依頼する際、治験実施計画書とともに治験薬概要書を提出します。医療機関では、治験審査委員会(IRB)が、治験薬概要書や治験実施計画書の内容を確認し、当該治験を実施してよいかどうかを審査します。IRBは、被験者に不当な危険が及ばないか、説明同意の内容は妥当か、観察や検査の計画は適切かといった観点から評価を行います。したがって、治験薬概要書は、現場の参考資料であるだけでなく、治験実施の可否を判断するうえでも重要な文書です。
また、治験責任医師や治験分担医師は、その内容を十分に理解したうえで治験を実施しなければなりません。治験中に副作用が疑われる症状が認められた場合にも、それが既知の事象か、新たなリスクの可能性があるかを判断するための重要な参考資料となります。CRCや治験ナースにとっても、被験者対応、来院時の確認事項、観察ポイントの把握などに役立つ実務文書です。さらに、治験依頼者が当局へ治験届書を提出する際にも、治験実施計画書と並んで重要な添付資料となります。
治験薬概要書の特徴の一つは、作成後も開発の進行に応じて改訂される点です。国内外の治験で新たな副作用が報告された場合、予期しない検査値異常が認められた場合、新たな有効性の知見が得られた場合、あるいは海外で規制上の取扱いが変更された場合などには、その内容を反映する必要があります。特に安全性に関する新情報は迅速な共有が求められ、改訂によって副作用への注意事項、観察ポイント、中止基準、併用注意などが見直されることがあります。これは被験者保護に直結するため、治験薬概要書は運用上も極めて重要な文書です。
CTDや薬事の観点から見ると、治験薬概要書は承認申請資料そのものではありませんが、承認申請に至るまでの開発過程で蓄積された非臨床及び臨床の知見を、治験現場で利用できる形に実務的に統合した文書です。治験を安全かつ科学的に進めるための基盤であり、治験実施計画書と並ぶ中心的文書の一つといえます。


Clinical Study
Investigator’s Brochure
The Investigator’s Brochure is a compilation of key information on the investigational product prepared for healthcare professionals involved in a clinical trial, including the investigator, subinvestigators, study nurses, and clinical research coordinators (CRCs). It is a structured summary of the clinical and non-clinical data obtained on the investigational product to date and serves as a basic reference for the proper conduct of the trial while ensuring the safety of the subjects.
Put simply for a general audience, the Investigator’s Brochure is a handbook that helps clinical trial personnel understand what kind of drug the investigational product is, what studies have been conducted so far, and what is known about its potential benefits and risks. Because an investigational product has not yet been approved and differs from drugs already used in routine medical practice, physicians and staff involved in the trial need to understand in advance its characteristics and the precautions associated with its use. The Investigator’s Brochure is the central source of such information.
Under Japan’s Good Clinical Practice (GCP), the sponsor is required to prepare an Investigator’s Brochure on the basis of materials obtained from prescribed studies, together with information on the quality, efficacy, and safety of the investigational product. At a minimum, the brochure must include the chemical name or development code of the investigational product, information on its quality, toxicity, pharmacology, and other relevant characteristics, and, where clinical studies have been conducted, the results of those studies. In practice, the brochure also systematically includes other information useful for subject management, such as the rationale for dosage and administration, known adverse reactions, clinically significant changes in laboratory values that require attention, precautions regarding concomitant medications, and risks suggested by preclinical studies.
When the sponsor requests a medical institution to conduct a clinical trial, the sponsor submits the Investigator’s Brochure together with the protocol. At the medical institution, the Institutional Review Board (IRB) reviews the contents of the Investigator’s Brochure and the protocol and determines whether the trial may appropriately be conducted. The IRB evaluates such matters as whether subjects may be exposed to unreasonable risk, whether the informed consent materials are appropriate, and whether the observation and examination plans are adequate. Accordingly, the Investigator’s Brochure is not merely a reference document for use at the study site, but also an important document in determining whether the trial itself may be conducted.
In addition, the investigator and subinvestigators must conduct the trial only after fully understanding the contents of the Investigator’s Brochure. If symptoms suggestive of an adverse reaction are observed during the trial, the brochure serves as an important reference in determining whether the event is already known or may represent a potential new risk. For CRCs and study nurses as well, it is a practical document that is useful for subject care, confirmation of items to be checked at study visits, and identification of key points for observation. Furthermore, when the sponsor submits a clinical trial notification to the regulatory authority, the Investigator’s Brochure is also included as an important attachment together with the protocol.
One important feature of the Investigator’s Brochure is that it is revised as development progresses. For example, revision may be required if a new adverse reaction is reported in a domestic or overseas clinical trial, if an unexpected laboratory abnormality is identified, if new evidence of efficacy is obtained, or if the regulatory treatment of the drug changes overseas. In particular, new safety information must be shared promptly, and revisions may lead to updates in adverse reaction warnings, observation points, discontinuation criteria, and precautions for concomitant use. Because such updates are directly linked to subject protection, the Investigator’s Brochure is an operationally critical document.
From the perspective of CTD and regulatory affairs, the Investigator’s Brochure is not itself part of the marketing application dossier. However, it is a practical document that integrates the non-clinical and clinical knowledge accumulated during development into a form that can be used at the clinical trial site. It is therefore one of the central documents, together with the protocol, that supports the safe and scientifically sound conduct of a clinical trial.
治験実施計画書
治験実施計画書は、治験をどのような目的で、どのような方法により、どのようなルールに従って実施するかを定めた基本文書です。治験責任医師、治験分担医師、治験ナース、治験コーディネーター(CRC)など、治験に関わる医療従事者が共通の手順で治験を進めるための中心的文書であり、治験全体の設計図に相当します。
内容としては、治験の目的、対象となる被験者、被験薬の用法・用量、観察及び検査項目、有効性及び安全性の評価方法、統計解析の考え方などがあらかじめ定められており、治験を科学的かつ倫理的に実施するための基盤となります。
わかりやすく言えば、治験実施計画書は「この治験をどのような決まりに従って進めるか」を示した詳しい手順書です。例えば、どのような患者さんに参加してもらうのか、どの薬をどのくらいの量でどのくらいの期間使うのか、どのような検査をいつ行うのか、どのような結果をもって効果を判断するのか、安全性に問題があった場合にどう対応するのか、といった事項が具体的に記載されます。治験は通常の診療とは異なり、薬の効果や安全性を客観的に確認するために行われるため、実施方法が施設や担当者によって異なっては正しい評価ができません。そのため、関係者全員が同じ基準で治験を実施できるように、治験実施計画書が作成されます。
日本の「医薬品の臨床試験の実施の基準」(GCP)では、治験依頼者に対し、治験ごとに適切な治験実施計画書を作成することが求められています。通常、治験の背景及び目的、試験デザイン、被験者の選択基準及び除外基準、併用禁止薬又は併用制限、観察及び検査項目、有害事象の取扱い、中止基準、統計解析方法などが記載されます。すなわち、治験実施計画書は、単なる予定表ではなく、「何を、なぜ、どのように評価するのか」を体系的に示す文書です。
治験実施計画書の重要な役割の一つは、治験の科学的妥当性を確保することです。対象となる患者集団、投与方法、評価時点、比較方法、解析方法などが適切に定められていなければ、得られた結果が薬の真の効果や安全性を示しているのか判断できません。治験実施計画書は、こうしたばらつきを可能な限り抑え、信頼性の高いデータを得るための条件を明確にする文書です。
また、治験実施計画書は被験者保護の観点からも非常に重要です。適格基準・除外基準、安全性確認のための検査スケジュール、有害事象発生時の対応、中止基準、緊急時の取扱いなどは、被験者の安全と権利を守るうえで欠かせません。このため、治験実施計画書は、薬の効果を調べるためだけではなく、被験者を適切に管理するための文書でもあります。
治験依頼者は、治験を医療機関に依頼する際、治験薬概要書とともに治験実施計画書を提出します。医療機関では、治験審査委員会(IRB)がその内容を確認し、当該治験を実施してよいかどうかを審査します。IRBは、治験の目的が妥当であるか、被験者に過大な危険や負担がないか、観察項目や来院頻度が適切か、説明同意文書と整合しているかなどを確認します。したがって、治験実施計画書は、現場で用いる運用文書であると同時に、治験の倫理性及び科学性を評価するための重要な審査資料でもあります。
治験現場では、治験責任医師、治験分担医師、CRC、治験ナースなどが、この治験実施計画書に従って被験者の登録、投与、観察及び評価を行います。計画書から外れた対応が行われた場合にはprotocol deviationとなることがあり、被験者保護やデータの信頼性に影響する可能性があります。そのため、治験実施計画書の遵守は極めて重要です。
さらに、治験実施計画書は開発の進行や新たな知見の入手に応じて改訂されることがあります。例えば、安全性情報の追加、対象被験者条件の見直し、評価方法の変更などが必要となった場合です。CTDや薬事の観点から見ると、治験実施計画書は承認申請資料そのものではありませんが、承認申請に用いられる臨床試験成績の質と信頼性を支える基盤文書です。治験を安全かつ科学的に進めるために、治験薬概要書と並んで中心的な役割を担う文書といえます。

Clinical Trial Protocol
A Clinical Trial Protocol is a fundamental document that specifies the purpose of a clinical trial, the methods by which it will be conducted, and the rules that must be followed throughout its conduct. It is a central document used by healthcare professionals involved in the trial, including the investigator, subinvestigators, study nurses, and clinical research coordinators (CRCs), to ensure that the trial is carried out according to common procedures. In that sense, it may be regarded as the blueprint for the entire clinical trial.
It sets out in advance such matters as the objectives of the trial, the subjects to be enrolled, the dosage and administration of the investigational product, the observations and examinations to be performed, the methods for evaluating efficacy and safety, and the statistical approach to be used. It therefore serves as the foundation for conducting the trial in a scientifically sound and ethically appropriate manner.
Put simply for a general audience, the Clinical Trial Protocol is a detailed instruction manual that explains the rules according to which the trial will be conducted. For example, it describes what kinds of patients may participate, which drug will be used, at what dose and for how long, what tests will be performed and when, how treatment effects will be judged, and how safety concerns will be handled if they arise. Because a clinical trial is conducted to evaluate the efficacy and safety of a drug objectively, unlike routine medical care, it is not acceptable for each institution or each investigator to proceed in a different way. For that reason, the Clinical Trial Protocol is prepared so that everyone involved can conduct the trial according to the same standards.
Under Japan’s Good Clinical Practice (GCP), the sponsor is required to prepare an appropriate Clinical Trial Protocol for each trial. The protocol generally includes the background and objectives of the trial, the study design, the inclusion and exclusion criteria for subjects, prohibited or restricted concomitant medications, the observation and examination items, the handling of adverse events, discontinuation criteria, and the statistical analysis methods. In other words, the Clinical Trial Protocol is not merely a timetable for the study; it is a document that systematically explains what will be evaluated, why it will be evaluated, and how that evaluation will be performed.
One of the major roles of the Clinical Trial Protocol is to ensure the scientific validity of the trial. Unless such matters as the target patient population, dosing method, evaluation time points, comparison methods, and analytical methods are appropriately defined, it cannot be determined whether the results obtained truly reflect the efficacy and safety of the drug. The Clinical Trial Protocol therefore clarifies the conditions necessary to minimize variability as much as possible and to obtain reliable data.
The Clinical Trial Protocol is also extremely important from the perspective of subject protection. Eligibility and exclusion criteria, the schedule of safety assessments, the handling of adverse events, discontinuation criteria, and emergency procedures are all essential for protecting the safety and rights of subjects. Accordingly, the Clinical Trial Protocol is not only a document for investigating the effects of a drug, but also a document for ensuring the proper management of subjects participating in the trial.
When the sponsor requests a medical institution to conduct the trial, the sponsor submits the Clinical Trial Protocol together with the Investigator’s Brochure. At the medical institution, the Institutional Review Board (IRB) reviews the contents of the protocol and determines whether the trial may appropriately be conducted. The IRB examines, for example, whether the objectives of the trial are appropriate, whether subjects will be exposed to excessive risk or burden, whether the observation items and visit frequency are suitable, and whether the protocol is consistent with the informed consent documents. Thus, the Clinical Trial Protocol serves not only as an operational document for use at the study site, but also as an important review document for evaluating the ethical and scientific acceptability of the trial.
At the trial site, the investigator, subinvestigators, CRCs, and study nurses carry out subject enrollment, dosing, observation, and evaluation in accordance with the Clinical Trial Protocol. If procedures are carried out outside the protocol, they may constitute a protocol deviation, which can affect subject protection and the reliability of the data. For that reason, compliance with the Clinical Trial Protocol is critically important.
In addition, the Clinical Trial Protocol may be revised as development progresses or as new information becomes available. For example, revision may be required when new safety information is obtained, when subject eligibility criteria need to be reconsidered, or when assessment methods need to be changed. From the perspective of CTD and regulatory affairs, the Clinical Trial Protocol is not itself part of the marketing application dossier, but it is a foundational document that supports the quality and reliability of the clinical trial data used in a marketing application. It may therefore be said, together with the Investigator’s Brochure, to play a central role in ensuring that the clinical trial is conducted safely and scientifically.
翻訳会社の強みを活かした メディカルライティング
Medical Writing Services That Utilize Our Strengths as a Translation Company

当社では医薬品申請文書の翻訳とメディカルライティングを同時並行で行うことができます。
また、クライアントのグローバル本社とのコレスポンデンスなどを含め、翻訳や文書作成プロジェクト全体の運営を日・英バイリンガルで支援することができます。
当社では、クライアントが翻訳・メディカルライティング業務での負担や時間的ロスを削減し、新薬開発と承認期間を短縮して頂けるよう、医薬品開発の各プロセスにおいて、迅速・正確な翻訳とメディカルライティン及びQC点検サービスを提供しております。
MedicaLingual can provide our clients with simultaneous approval application translation and medical writing services.
MedicaLingual can also provide bilingual (Japanese/English) support throughout the entire translation and document preparation project, including help with correspondence between the Japan and global teams.
MedicaLingual provides its clients with prompt and accurate translation, medical writing, and QC inspection services throughout every step of the drug development process to make it possible for them to reduce their workload and save time on translation and medical writing and thereby shorten the time it takes for new drug development and approval.





