

薬生薬審発0202第1号(平成29年2月2日)
ICH HARMONISED GUIDELINE- M4E(R2) – Dated 15 June 2016
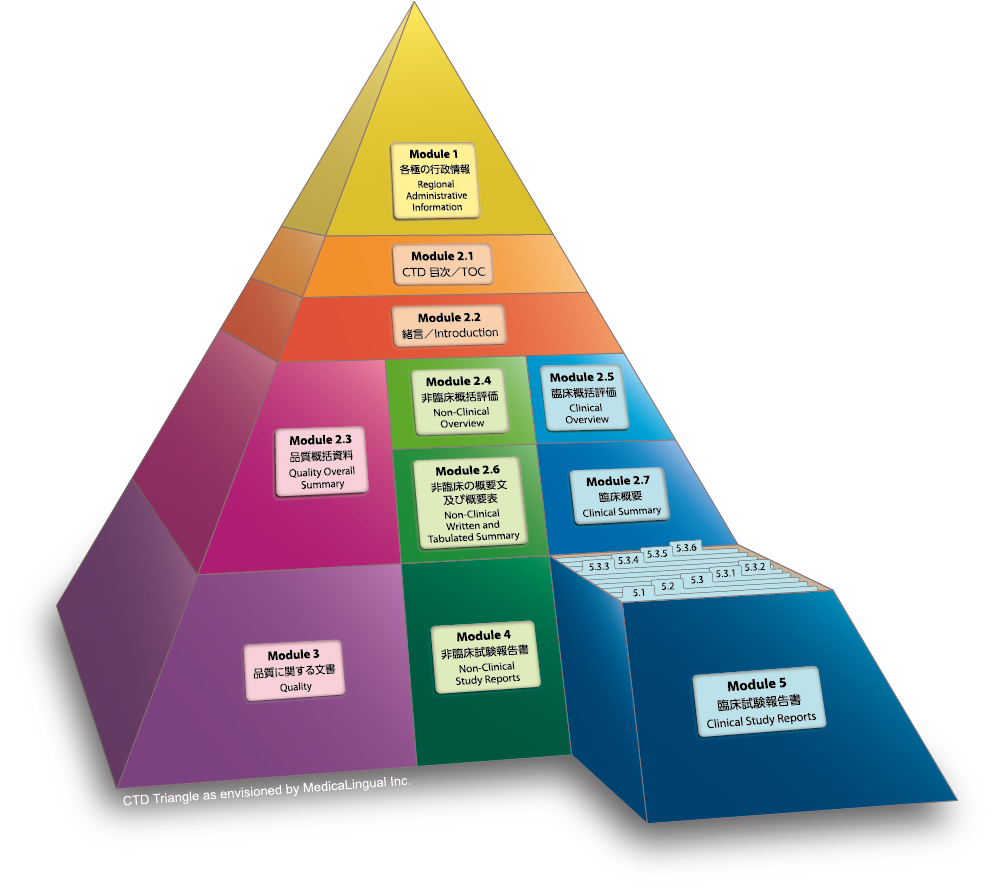
Module 5 臨床試験報告書
序文
「治験の総括報告書の構成と内容に関するガイドライン(E3)」が公表されているが、この第5部では、医薬品承認申請のための国際共通化資料(CTD)における総括報告書、その他臨床データ及び参考文献の配列に関する指針を示すものである。これらCTD資料の配列は、承認申請書類の作成及び当局による審査を容易にするものである。
このガイドラインは、臨床試験報告書の配列を示すものであり、承認取得のためにどのような試験が要求されるのかを示すものではない。
Module 5 CLINICAL STUDY REPORTS
Preamble
Through the ICH process, a guideline has been published on the structure and content of clinical study reports (E3). This document provides guidance on the organisation of these study reports, other clinical data, and references within a Common Technical Document (CTD) for registration of a pharmaceutical product for human use. These elements should facilitate the preparation and review of a marketing application.
This guideline is not intended to indicate what studies are required for successful registration. It indicates an appropriate organization for the clinical study reports that are in the application.
第 5 部(モジュール 5)における臨床試験報告書 及び関連情報の詳細な配列
本ガイドラインは、承認申請書類の作成とその審査が円滑に行われるよう、また資料を完全なものにするため、臨床試験報告書及び関連情報の一定の配列を推奨するものである。報告書の配列は、主たる試験目的によって決めること。各報告書は一つの項のみに添付し、複数目的の場合は、添付を繰返さずその項を参照すること。添付する報告書又は情報がない項目には、「該当せず」又は「試験を実施せず」等と記載すること。
Detailed Organisation of Clinical Study Reports and Related Information in Module 5.
This guideline recommends a specific organization for the placement of clinical study reports and related information to simplify preparation and review of dossiers and to ensure completeness. The placement of a report should be determined by the primary objective of the study. Each study report should appear in only one section. Where there are multiple objectives, the study should be cross-referenced in the various sections. An explanation such as “not applicable” or “no study conducted” should be provided when no report or information is available for a section or subsection.
5.1 目次
試験報告書の目次を作成すること。
5.1 Table of Contents of Module 5
A Table of Contents for study reports should be provided.
5.2 臨床試験一覧表
全ての臨床試験及び関連情報の一覧表を作成すること。試験ごとの記載内容は、通常、本ガイドラインの表5.1に示す情報が含まれることになる。その他の情報は、申請者が有用と判断するものを含めてよい。順序は、下記第5.3項の配列に従うこと。異なる配列を使用する場合は、一覧表の序文にその旨を説明すること。
5.2 Tabular Listing of All Clinical Studies
A tabular listing of all clinical studies and related information should be provided. For each study, this tabular listing should generally include the type of information identified in Table 5.1 of this guideline. Other information can be included in this table if the applicant considers it useful. The sequence in which the studies are listed should follow the sequence described in Section 5.3 below. Use of a different sequence should be noted and explained in an introduction to the tabular listing.
5.3 試験報告書及び関連情報
5.3.1 生物薬剤学試験報告書
BA 試験は、医薬品から有効成分が放出される速度と程度を評価するものである。比較 BA 試験又は BE 試験は、PK、PD、臨床的又は in vitro の溶出性等のエンドポイントを用い、単回又は反復投与で実施される。試験の主たる目的は PK の評価であるが、同時に BA 情報を得たものであれば、その結果は第 5.3.1 項に添付し、第 5.3.1.1 項、第5.3.1.2 項では第 5.3.1 項を参照すること。
5.3 Clinical Study Reports
5.3.1 Reports of Biopharmaceutic Studies
BA studies evaluate the rate and extent of release of the active substance from the medicinal product. Comparative BA or BE studies may use PK, PD, clinical, or in vitro dissolution endpoints, and may be either single dose or multiple dose. When the primary purpose of a study is to assess the PK of a drug, but also includes BA information, the study report should be submitted in Section 5.3.1, and referenced in Sections 5.3.1.1 and/or 5.3.1.2.
5.3.1.1 バイオアベイラビリティ(BA)試験報告書
本項の BA 試験には以下の試験が含まれる。
- 経口固形製剤からの有効成分の放出及び全身的利用率を、静脈内投与又は経口液剤による全身的利用率と比較した試験
- 含量の異なる製剤間における比例性試験
- 食事の影響に関する試験
5.3.1.1 Bioavailability (BA) Study Reports
BA studies in this section should include
- studies comparing the release and systemic availability of a drug substance from a solid oral dosage form to the systemic availability of the drug substance given intravenously or as an oral liquid dosage form
- dosage form proportionality studies, and
- food-effect studies.
5.3.1.2 比較 BA 試験及び生物学的同等性(BE)試験報告書
本項に添付する試験は、有効成分放出の速度と程度を類似した製剤間(例:錠剤と錠剤、錠剤とカプセル)で比較しているものとする。比較 BA 又は BE 試験に含まれるものとしては以下の試験があげられる。
- 有効性を裏付ける試験で用いられた製剤と市販予定製剤との比較試験
- 有効性を裏付ける試験で用いられた製剤と安定性試験で用いられた製剤との比較試験
- 製造者が異なる製剤間の比較試験
5.3.1.2 Comparative BA and Bioequivalence (BE) Study Reports
Studies in this section compare the rate and extent of release of the drug substance from similar drug products (e.g., tablet to tablet, tablet to capsule). Comparative BA or BE studies may include comparisons between
- the drug product used in clinical studies supporting effectiveness and the to-be-marketed drug product,
- the drug product used in clinical studies supporting effectiveness and the drug product used in stability batches, and
- similar drug products from different manufacturers.
5.3.1.3 In Vitro-In Vivo の関連を検討した試験報告書
In vivo との関連を検討した in vitro 試験等、BA に関する情報が得られた in vitro 溶出試験があれば、この第 5.3.1.3 項に添付すること。バッチの品質管理、バッチの出庫可否判断のために用いた in vitro 溶出試験報告書は、CTD の第 3 部(モジュール 3)「品質に関する文書」に添付すること。
5.3.1.3 In Vitro – In Vivo Correlation Study Reports
In vitro dissolution studies that provide BA information, including studies used in seeking to correlate in vitro data with in vivo correlations, should be placed in Section 5.3.1.3. Reports of in vitro dissolution tests used for batch quality control and/or batch release should be placed in the Quality section of the CTD.
5.3.1.4 生物学的及び理化学的分析法検討報告書
生物薬剤学試験又は in vitro 溶出試験で用いた生物学的分析法、理化学的分析法は、通常、個々の試験報告書に記載すること。ある一つの方法が複数試験で用いられた場合は、方法及びそのバリデーションに関する報告書を第 5.3.1.4 項に添付し、個々の試験報告書においてはそれを引用すること。
5.3.1.4 Reports of Bioanalytical and Analytical Methods for Human Studies
Bioanalytical and/or analytical methods for biopharmaceutic studies or in vitro dissolution studies should ordinarily be provided in individual study reports. Where a method is used in multiple studies, the method and its validation should be included once in Section 5.3.1.4 and referenced in the appropriate individual study reports.
5.3.2 ヒト生体試料を用いた薬物動態関連の試験報告書
ヒト生体試料とは、有効成分のPK特性を評価するためにin vitroやex vivoで用いられたヒトに由来する蛋白質、細胞、組織、及び関連試料を指す用語である。例として、生体膜透過性及び輸送過程の評価のために用いられる培養ヒト結腸細胞や血漿蛋白結合検討のために用いられるヒトアルブミン等がある。特に重要なものは、薬物代謝経路を検討し、代謝経路に関係する薬物-薬物相互作用を検討するための肝細胞、肝ミクロソーム等のヒト生体試料である。その他の特性(例:無菌性や薬力学)を検討するために生体試料を用いて実施した試験は、臨床試験報告書の部に添付せず、非臨床試験報告書の部に添付すること(第4部(モジュール4))。
5.3.2 Reports of Studies Pertinent to Pharmacokinetics Using Human Biomaterials
Human biomaterials is a term used to refer to proteins, cells, tissues and related materials derived from human sources that are used in vitro or ex vivo to assess PK properties of drug substances. Examples include cultured human colonic cells that are used to assess permeability through biological membranes and transport processes, and human albumin that is used to assess plasma protein binding. Of particular importance is the use of human biomaterials such as hepatocytes and/or hepatic microsomes to study metabolic pathways and to assess drug-drug interactions with these pathways. Studies using biomaterials to address other properties (e.g., sterility or pharmacodynamics) should not be placed in the Clinical Study Reports Section, but in the Nonclinical Study Section (Module 4).
5.3.2.1 血漿蛋白結合試験報告書
Ex vivo蛋白結合試験の報告書は、本項に添付すること。PK試験から得られた蛋白結合に関するデータは、第3項に添付すること。
5.3.2.1 Plasma Protein Binding Study Reports
Ex vivo protein binding study reports should be provided here. Protein binding data from PK blood and/or plasma studies should be provided in Section 5.3.3.
5.3.2.2 肝代謝及び薬物相互作用試験報告書
肝組織を用いた代謝及び薬物相互作用試験の報告書は、本項に添付すること。
5.3.2.2 Reports of Hepatic Metabolism and Drug Interaction Studies
Reports of hepatic metabolism and metabolic drug interaction studies with hepatic tissue should be placed here.
5.3.2.3 他のヒト生体試料を用いた試験報告書
その他の生体試料を用いた試験の報告書は、本項に添付すること。
5.3.2.3 Reports of Studies Using Other Human Biomaterials
Reports of studies with other biomaterials should be placed in this section.
5.3.3 臨床薬物動態(PK)試験報告書
健康被験者、患者におけるPK特性の評価は、投与計画及び用量調整の検討や、併用薬剤による影響の予測、観察された薬力学的作用の差異を解釈する上で重要である。PK評価は、親化合物及び代謝物(特に薬理活性がある場合)が、生体内でどのような挙動を示すかを経時的に検討するものであり、最高血漿中濃度(最大曝露量)、AUC(総曝露量)、クリアランス、そして蓄積に焦点が置かれるものである。
第5.3.3.1項及び第5.3.3.2項の対象となるPK試験の目的としては、(1)血漿中薬物及び代謝物濃度の経時的測定、(2)有用又は必要な場合には尿中又は糞中の薬物及び代謝物濃度の測定、(3)蛋白質や赤血球と結合した薬物及び代謝物の測定等が一般的である。
PK試験には、組織、臓器、体液(例:滑液や脳脊髄液)への薬物分布測定が含まれる場合もあるが、その結果は第5.3.3.1項又は第5.3.3.2項に適切に添付すること。これらの試験により、薬物のPK特性を明らかにするとともに、健康被験者、患者における薬物及び活性代謝物の吸収・分布・代謝・排泄に関する情報を提供すること。マスバランス試験や、用量(例:用量比例性)や時間(例:酵素誘導又は抗体産生の影響)と関連したPKの変化を検討した試験は特に重要であり、結果は第5.3.3.1項又は第5.3.3.2項に添付すること。健康志願者及び患者におけるPKの測定結果は、平均値による記述とは別に、個々の変動範囲についても記述すること。外国臨床データを受け入れる際に考慮すべき民族的要因に関するICH E5ガイドラインでは、異なる集団における異なる薬物反応をもたらす要因として内因性民族的要因と外因性民族的要因を挙げている。本ガイドラインでは、これらの分類をそれぞれ内因性要因及び外因性要因と記述する。これら内因性要因(例:年齢、性、人種、体重、身長、疾患、遺伝子多型及び臓器機能不全)及び外因性要因(例:薬物間相互作用、食事、喫煙及び飲酒)によるPKの変化の結果としての全身曝露量の違いを評価することもある。これら内因性及び外因性要因の影響を検討した試験報告書は、第5.3.3.3項及び第5.3.3.4項にそれぞれ添付すること。
繰返しサンプルを採取する標準的PK試験に加え、臨床試験において少数回サンプルを採取するポピュレーションPK解析によっても、用量-PK-反応関係の変化における内因性及び外因性要因の関与を検討することができる。ポピュレーションPK試験で用いられる方法は、標準的PK試験で用いられるものとは本質的に異なっているため、これらの試験報告書は第5.3.3.5項に添付すること。
5.3.3 Reports of Human Pharmacokinetic (PK) Studies
Assessment of the PK of a drug in healthy subjects and/or patients is considered critical to designing dosing strategies and titration steps, to anticipating the effects of concomitant drug use, and to interpreting observed pharmacodynamic differences. These assessments should provide a description of the body’s handling of a drug over time, focusing on maximum plasma concentrations (peak exposure), area-under-curve (total exposure), clearance, and accumulation of the parent drug and its metabolite(s), in particular those that have pharmacological activity.
The PK studies whose reports should be included in Sections 5.3.3.1 and 5.3.3.2 are generally designed to (1) measure plasma drug and metabolite concentrations over time, (2) measure drug and metabolite concentrations in urine or faeces when useful or necessary, and/or (3) measure drug and metabolite binding to protein or red blood cells.
On occasion, PK studies may include measurement of drug distribution into other body tissues, body organs, or fluids (e.g., synovial fluid or cerebrospinal fluid), and the results of these tissue distribution studies should be included in Section 5.3.3.1 to 5.3.3.2, as appropriate. These studies should characterise the drug’s PK and provide information about the absorption, distribution, metabolism, and excretion of a drug and any active metabolites in healthy subjects and/or patients. Studies of mass balance and changes in PK related to dose (e.g., determination of dose proportionality) or time (e.g., due to enzyme induction or formation of antibodies) are of particular interest and should be included in Sections 5.3.3.1 and/or 5.3.3.2. Apart from describing mean PK in normal and patient volunteers, PK studies should also describe the range of individual variability. In the ICH E5 guideline on Ethnic Factors in the Acceptance of Foreign Data, factors that may result in different responses to a drug in different populations are categorised as intrinsic ethnic factors or extrinsic ethnic factors. In this document, these categories are referred to as intrinsic factors and extrinsic factors, respectively. Additional studies can also assess differences in systemic exposure as a result of changes in PK due to intrinsic (e.g., age, gender, racial, weight, height, disease, genetic polymorphism, and organ dysfunction) and extrinsic (e.g., drug-drug interactions, diet, smoking, and alcohol use) factors. Reports of PK studies examining the influence of intrinsic and extrinsic factors on exposure should be organised in Sections 5.3.3.3 and 5.3.3.4, respectively.
In addition to standard multiple-sample PK studies, population PK analyses based on sparse sampling during clinical studies can also address questions about the contributions of intrinsic and extrinsic factors to the variability in the dose-PK-response relationship. Because the methods used in population PK studies are substantially different from those used in standard PK studies, these studies should be placed in Section 5.3.3.5.
5.3.3.1 健康被験者におけるPK及び初期忍容性試験報告書
健康被験者におけるPK及び初期忍容性試験の報告書は、本項に添付すること。
5.3.3.1 Healthy Subject PK and Initial Tolerability Study Reports
Reports of PK and initial tolerability studies in healthy subjects should be placed in this section.
5.3.3.2 患者におけるPK及び初期忍容性試験報告書
患者におけるPK及び初期忍容性試験の報告書は、本項に添付すること。
5.3.3.2 Patient PK and Initial Tolerability Study Reports
Reports of PK and initial tolerability studies in patients should be placed in this section.
5.3.3.3 内因性要因を検討したPK試験報告書
内因性要因を検討したPK試験報告書は、本項に添付すること。
5.3.3.3 Intrinsic Factor PK Study Reports
Reports of PK studies to assess effects of intrinsic factors, should be placed in this section.
5.3.3.4 外因性要因を検討したPK試験報告書
外因性要因を検討したPK試験報告書は、本項に添付すること。
5.3.3.4 Extrinsic Factor PK Study Reports
Reports of PK studies to assess effects of extrinsic factors, should be placed in this section.
5.3.3.5 ポピュレーションPK試験報告書
有効性及び安全性試験を含む臨床試験で少数回採取したサンプルに基づくポピュレーションPK試験の報告書は、本項に添付すること。
5.3.3.5 Population PK Study Reports
Reports of population PK studies based on sparse samples obtained in clinical trials including efficacy and safety trials, should be placed in this section.
5.3.4 臨床薬力学(PD) 試験報告書
ヒトにおける医薬品のPD作用を検討することを主目的とする試験の報告書は、本項に添付すること。しかし、有効性の確立や、安全性データの集積が主目的である場合には、第5.3.5項に添付すること。
本項の対象となる試験には、(1)期待される臨床効果と関連するか関連すると考えられる薬理学的特性(生物学的マーカー)に関する試験、(2)主たる臨床的効果を検討する短期試験、(3)期待される臨床効果と関連しないその他の特性に関するPD試験等がある。薬理学的効果と用量ないし血漿中薬物/代謝物濃度との定量的関係については、通常関心のあるところであり、PD関連情報は、用量反応試験あるいはPK試験(濃度-反応又はPK/PD試験)における薬物濃度に関する情報と共に得られることが多い。適切に管理された試験から得られたものではないPKとPD作用との関係は、しばしばモデルを用いて評価され、用量反応試験のデザインの根拠として、時に、部分集団における薬物濃度の違いの影響を解釈する根拠として利用される。
用量探索試験、PD試験、PK-PD試験は、健康被験者及び/又は患者を対象に実施されるが、それらの試験を、ある適応症を対象に行う安全性及び有効性試験に組み入れることもできる。健康被験者で実施されたこれらの試験報告書は第5.3.4.1項に、患者で実施された報告書は第5.3.4.2項に添付すること。
患者におけるPD試験で得られる短期PD、用量探索、PK-PDに関する情報には、受け入れられる代用マーカー(例:血圧)や治療上のベネフィットを示すエンドポイント(例:疼痛の緩和)に対する効果を反映するものもあるため、有効性評価に寄与するデータを提供する場合もある。同様に、PD試験が重要な安全性情報を含むこともある。ゆえに、PD試験のうち有効性や安全性を証明するものがあれば、その試験は有効性及び安全性試験とみなし、第5.3.4項ではなく第5.3.5項に添付すること。
5.3.4 Reports of Human Pharmacodynamic (PD) Studies
Reports of studies with a primary objective of determining the PD effects of a drug product in humans should be placed in this section. Reports of studies whose primary objective is to establish efficacy or to accumulate safety data, however, should be placed in Section 5.3.5.
This section should include reports of 1) studies of pharmacologic properties known or thought to be related to the desired clinical effects (biomarkers), 2) short-term studies of the main clinical effect, and 3) PD studies of other properties not related to the desired clinical effect. Because a quantitative relationship of these pharmacological effects to dose and/or plasma drug and metabolite concentrations is usually of interest, PD information is frequently collected in dose response studies or together with drug concentration information in PK studies (concentration-response or PK/PD studies). Relationships between PK and PD effects that are not obtained in well-controlled studies are often evaluated using an appropriate model and used as a basis for designing further dose-response studies or, in some cases, for interpreting effects of concentration differences in population subsets.
Dose-finding, PD and/or PK-PD studies can be conducted in healthy subjects and/or patients, and can also be incorporated into the studies that evaluate safety and efficacy in a clinical indication. Reports of dose-finding, PD and/or PK/PD studies conducted in healthy subjects should be placed in Section 5.3.4.1, and the reports for those studies conducted in patients should be placed in Section 5.3.4.2.
In some cases, the short-term PD, dose-finding, and/or PK-PD information found in pharmacodynamic studies conducted in patients will provide data that contribute to assessment of efficacy, either because they show an effect on an acceptable surrogate marker (e.g., blood pressure) or on a clinical benefit endpoint (e.g., pain relief). Similarly, a PD study may contain important clinical safety information. When these studies are part of the efficacy or safety demonstration, they are considered clinical efficacy and safety studies that should be included in Section 5.3.5, not in Section 5.3.4.
5.3.4.1 健康被験者におけるPD試験及びPK/PD試験報告書
健康被験者を対象とした、治療を目的としないPD試験、PK/PD試験は、本項に添付すること。
5.3.4.1 Healthy Subject PD and PK/PD Study Reports
PD and/or PK/PD studies having non-therapeutic objectives in healthy subjects should be placed in this section.
5.3.4.2 患者におけるPD試験及びPK/PD試験報告書
患者を対象としたPD試験、PK/PD試験は、本項に添付すること。
5.3.4.2 Patient PD and PK/PD Study Reports
PD and/or PK/PD studies in patients should be submitted in this section.
5.3.5 有効性及び安全性試験報告書
本項には、申請医薬品の有効性、安全性に関する全ての臨床試験報告書を添付すること。対象となるのは、治験依頼者が実施した試験か入手可能な報告書であり、申請適応症であるか否かを問わず、完了しているか進行中であるかを問わない。報告書の詳細さは、その試験の重要性及び申請における位置付けに相応したものであること。安全性と有効性の両方を裏付ける試験の報告書の内容についてはICH E3に記述されている。試験によっては、簡略化された報告書を提出してもよい(ICH E3及び地域ごとのガイダンスを参照)。
本項における配列は、試験のデザイン(比較対照試験、非対照試験)に拠ること。また比較対照試験の配列は、対照の種類に拠ること。さらに各項では、総括報告書と簡略化された報告書(ICH E3)とに分け、総括報告書を先に添付すること。データが限られているか、又は追加データが入手できない公表文献は、最後に添付すること。
複数の適応症を申請する場合には、適応症ごとに第5.3.5項を起こし、該当する報告書を分けて添付すること。ある有効性試験がひとつの適応症のみと関連している場合は、該当する適応症の第5.3.5項に添付すること。複数の適応症と関連している場合は、最も適切と考えられる適応症の第5.3.5項に添付し、必要に応じて他の適応症の第5.3.5項(例:第 5.3.5 肺炎、第 5.3.5 尿路感染症)においてはそれを参照すること。
5.3.5 Reports of Efficacy and Safety Studies
This section should include reports of all clinical studies of efficacy and/or safety carried out with the drug, conducted by the sponsor, or otherwise available, including all completed and all ongoing studies of the drug in proposed and non-proposed indications. The study reports should provide the level of detail appropriate to the study and its role in the application. ICH E3 describes the contents of a full report for a study contributing evidence pertinent to both safety and efficacy. Abbreviated reports can be provided for some studies (see ICH E3 and individual guidance by region).
Within Section 5.3.5, studies should be organised by design (controlled, uncontrolled) and, within controlled studies, by type of control. Within each section, studies should be categorized further, ordered by whether the study report is complete or abbreviated (ICH E3), with completely reported studies presented first. Published reports with limited or no further data available to the sponsor should be placed last in this section.
In cases where the application includes multiple therapeutic indications, the reports should be organized in a separate Section 5.3.5 for each indication. In such cases, if a clinical efficacy study is relevant to only one of the indications included in the application, it should be included in the appropriate Section 5.3.5; if a clinical efficacy study is relevant to multiple indications, the study report should be included in the most appropriate Section 5.3.5 and referenced as necessary in other Sections 5.3.5, e.g., Section 5.3.5A, Section 5.3.5B.
5.3.5.1 申請する適応症に関する比較対照試験報告書
比較対照試験報告書は、対照の種類により配列すること。
- プラセボ対照(実対照薬や他の用量等プラセボ以外の対照を同時に比較していてもよい)
- 無治療対照
- 用量反応対照(プラセボ対照なし)
- 実薬対照(プラセボ対照なし)
- 外部(既存)対照(対照治療にかかわらず)
薬剤の有効性評価と関連がある場合には、対照の種類別に、試験を投与期間ごとに添付すること。申請した適応症に関する試験ではないが、申請した用法・用量での有効性を裏付けるデータを提供する試験は、第5.3.5.1項に添付すること。
薬力学試験が有効性を裏付ける場合には、第5.3.5.1項に添付すること。試験が実施された時期的な順序は添付する順序には関連しない。したがって、プラセボ対照比較試験は、実施された時期にかかわらず、第5.3.5.1項に添付すること。安全性に関する比較対照試験も、申請の対象ではない条件での試験も含めて、第5.3.5.1項に添付すること。
5.3.5.1 Study Reports of Controlled Clinical Studies Pertinent to the Claimed Indication
The controlled clinical study reports should be sequenced by type of control:
- Placebo control (could include other control groups, such as an active comparator or other doses)
- No-treatment control
- Dose-response (without placebo)
- Active control (without placebo)
- External (Historical) control, regardless of the control treatment
Within each control type, where relevant to assessment of drug effect, studies should be organized by treatment duration. Studies of indications other than the one proposed in the application, but that provide support for efficacy in the proposed use, should be included in Section 5.3.5.1.
Where a pharmacodynamic study contributes to evidence of efficacy, it should be included in Section 5.3.5.1. The sequence in which studies were conducted is not considered pertinent to their presentation. Thus, placebo-controlled trials, whether early or late, should be placed in Section 5.3.5.1. Controlled safety studies, including studies in conditions that are not the subject of the application, should also be reported in Section 5.3.5.1.
5.3.5.2 非対照試験報告書
非対照試験の報告書(例:非盲検の安全性試験)は、本項に添付すること。これには承認申請の対象ではないものも含まれる。
5.3.5.2 Study Reports of Uncontrolled Clinical Studies
Study reports of uncontrolled clinical studies (e.g., reports of open label safety studies) should be included in Section 5.3.5.2. This includes studies in conditions that are not the subject of the marketing application.
5.3.5.3 複数の試験成績を併せて解析した報告書
申請に関する臨床上の問題点は、複数の試験成績を考慮した解析によって扱われることがある。そのような解析結果は、一般に臨床概要に要約されるべきであるが、解析結果を詳細に記述し提示することは、その結果を解釈する上で重要である。臨床概要に記載するには解析が詳細すぎるような場合には、それらを別の報告書としてまとめること。このような報告書は、第5.3.5.3項に添付すること。本項に含まれる報告書の例には、以下のものが含まれる。
- 全ての患者及び/又は特殊な部分集団における有効率の全体的な推定値を決定するための正式なメタアナリシス又は有効性に関する広範な探索的解析の報告書
- 安全性データベースの妥当性、有害事象発現率の推定、並びに用量、人口統計学的特性、及び併用薬といった変数に関連した安全性等を評価するための安全性に関する統合された解析の報告書
正式なブリッジング試験、関連する他の臨床試験、他の適切な情報(例:PK及びPD情報)を考慮したブリッジングに関する詳細な解析報告書は、臨床概要に含めるには長すぎる場合には、本項に添付すること。
5.3.5.3 Reports of Analyses of Data from More than One Study
Many clinical issues in an application can be addressed by an analysis considering data from more than one study. The results of such an analysis should generally be summarized in the clinical summary documents, but a detailed description and presentation of the results of such analyses are considered critical to their interpretation. Where the details of the analysis are too extensive to be reported in a summary document, they should be presented in a separate report. Such reports should be placed in Section 5.3.5.3. Examples of reports that would be found in this section include: a report of a formal meta-analysis or extensive exploratory analysis of efficacy to determine an overall estimate of effect size in all patients and/or in specific subpopulations, and a report of an integrated analysis of safety that assesses such factors as the adequacy of the safety database, estimates of event rates, and safety with respect to variables such as dose, demographics, and concomitant medications.
A report of a detailed analysis of bridging, considering formal bridging studies, other relevant clinical studies, and other appropriate information (e.g., PK and PD information), should be placed in this section if the analysis is too lengthy for inclusion in the Clinical Summary.
5.3.5.4 その他の臨床試験報告書
本項には以下の報告書が添付される。
- 申請された適応症に関連した試験の中間解析に関する報告書
- 他に報告されていない安全性に関する比較対照試験の報告書
- 申請された適応症とは関連しない比較対照試験又は非対照試験の報告書
- 第5.3.5.1項には含まれない、当該医薬品の臨床経験に関する公表文献。ただし、有効性の裏付けのために重要である場合には、第5.3.5.1項に添付すること。
- 進行中の試験に関する報告書
- 第5部の他の項に添付することが適当でないと判断された報告書(例:第2部第2.7.2.4項特別な試験)
5.3.5.4 Other Study Reports
This section can include:
- Reports of interim analyses of studies pertinent to the claimed indications
- Reports of controlled safety studies not reported elsewhere
- Reports of controlled or uncontrolled studies not related to the claimed indication
- Published reports of clinical experiences with the medicinal product that are not included in Section 5.3.5.1. However, when literature is important to the demonstration or substantiation of efficacy, it should be included in Section 5.3.5.1
- Reports of ongoing studies
5.3.6 市販後の使用経験に関する報告書
現在市販されている製品については、市販後の使用経験(全ての重要な安全性に関する知見を含む)をまとめた報告書を第5.3.6項に添付すること。
5.3.6 Reports of Post-Marketing Experience
For products that are currently marketed, reports that summarize marketing experience (including all significant safety observations) should be included in Section 5.3.6.
5.4 参考文献
重要な公表論文、公式な会議録、又は他の規制当局のガイダンスや助言を含む参考資料のコピーを本項に添付する。これには、臨床概括で引用した全ての参考文献のコピー、臨床概要又は第5部第5.3項に添付される個々の報告書で引用された重要な参考文献のコピーが含まれる。各参考文献についてはコピーを一部添付すること。本項に含まれない参考文献のコピーは、要求があれば直ちに提供すること。
5.4 Literature References
Copies of referenced documents, including important published articles, official meeting minutes, or other regulatory guidance or advice should be provided here. This includes copies of all references cited in the Clinical Overview, and copies of important references cited in the Clinical Summary or in the individual technical reports that were provided in Module 5, section 5.3. Only one copy of each reference should be provided. Copies of references that are not included here should be immediately available on request.





