

医薬審発第899号 CTD通知(別紙 3)
CTD-品質に関する文書の作成要領に関するガイドライン
(2002年9月11-12日ワシントン会議修正版)
ICH HARMONISED GUIDELINE
QUALITY – M4Q –
(Numbering and Section Headers have been edited for consistency and use in e-CTD as agreed at the Washington DC Meeting, September 11-12, 2002)
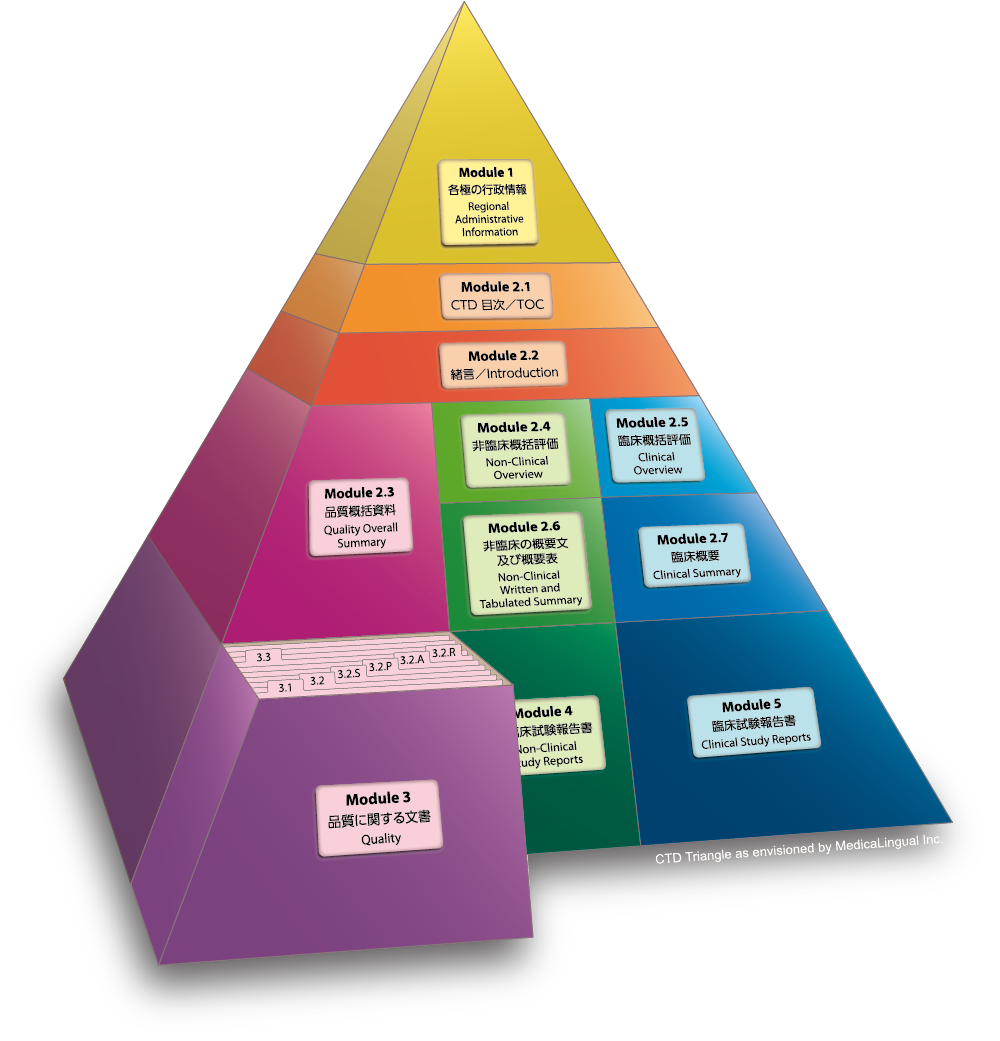
Module 3 品質に関する文書
適用範囲
本ガイドラインは、ICHガイドラインQ6A(以下、「新規化学薬品」と言う。)及びQ6B(バイオテクノロジー応用医薬品/生物起源由来医薬品。以下、「生物薬品」と言う。)の適用範囲において定義される原薬及びその製剤に係る申請資料の様式(項目と配列順序)に関する指針を示すことを目的とするものである。本ガイドラインに定める様式は上記以外の医薬品に適用可能な場合もあるので、それらの医薬品の申請にあたって、申請者は、本ガイドラインの適用の可否について規制当局に相談の上、その適用の可否を判断すること。
各項の表題に続く文章は、その項の内容を説明したり、明確にしたものにすぎない。各項の内容は、既存のICHガイドラインの記述に沿ったものとなっているが、既存のICHガイドラインがすべての内容を網羅しているわけではない。本ガイドラインの“データ又は報告書”中に記載された項目は、単に各資料の配列順序を示したものにすぎない。本ガイドラインには特定の必要なデータの種類や程度については規定されておらず、各極の方針に依存するものである。
3.2.R項(各極の要求資料)とは、3極に共通しない必要資料の代表的な例を示すものである。したがって、3.2.R項に挙げられるべき必要資料は各極の規制ガイドラインに依存している。
Module 3 QUALITY
SCOPE OF THE GUIDELINE
This document is intended to provide guidance on the format of a registration application for drug substances and their corresponding drug products as defined in the scope of the ICH Guidelines Q 6 A (“NCE”) and ICH Guideline Q 6 B (“Biotech”). This format may also be appropriate for certain other categories of products. To determine the applicability of this format for a particular type of product, applicants should consult with the appropriate regulatory authorities.
The text following the section titles is intended to be explanatory and illustrative only. The content of these sections should include relevant information described in existing ICH guidelines, but harmonised content is not available for all sections. The “Body of Data” in this guideline merely indicates where the information should be located. Neither the type nor extent of specific supporting data has been addressed in this guideline, and both may depend upon regional guidance.
The section titles of Part 3.2.R (Regional Information) represent examples of typical topics of information that are not common to all ICH regions. Hence, the information to be provided in these sections should be based on the relevant regional guidelines.
3.1 目次
添付資料の一覧表を作成する。
3.1 Table of Contents of Module 3
A Table of Contents for the filed application should be provided.
3.2 データ
3.2.S 原薬
複数の原薬を含む製剤にあっては、各原薬ごとに3.2.S項の資料を作成する。
3.2 Body of Data
3.2.S Drug Substance
For a drug product containing more than one drug substance, the information requested for part “S” should be provided in its entirety for each drug substance.
3.2.S.1 一般情報
3.2.S.1.1 名称
原薬の名称に関する事項を記述する。
記述する事項は、国際一般名(r-INN)、公定書収載名(もしあれば)、化学名、企業コード又は研究所コード、その他の一般名(BAN、USAN、JAN等の国ごとの名称等)、ケミカル・アブストラクト・サービス(CAS)登録番号等である。
3.2.S.1 General Information
3.2.S.1.1 Nomenclature
Information on the nomenclature of the drug substance should be provided. For example:
- Recommended International Nonproprietary Name (INN);
- Compendial name if relevant;
- Chemical name(s);
- Company or laboratory code;
- Other non-proprietary name(s), e.g., national name, United States Adopted Name
- (USAN), Japanese Accepted Name (JAN); British Approved Name (BAN), and
- Chemical Abstracts Service (CAS) registry number.
3.2.S.1.2 構造
新規化学薬品:
構造式(相対的及び絶対的立体化学を含む。)、分子式及び分子量を示す。
生物薬品:
適宜、糖鎖結合部位、その他翻訳後修飾を示したアミノ酸配列及び相対分子量を記載する。
3.2.S.1.2 Structure
NCE:
The structural formula, including relative and absolute stereochemistry, the molecular formula, and the relative molecular mass should be provided.
Biotech:
The schematic amino acid sequence indicating glycosylation sites or other posttranslational modifications and relative molecular mass should be provided, as appropriate.
3.2.S.1.3 一般特性
原薬の物理的化学的性質その他の適切な特性(生物薬品の場合には、生物活性を含む。)の一覧表を示す。
3.2.S.1.3 General Properties
A list should be provided of physicochemical and other relevant properties of the drug substance, including biological activity for Biotech.
Reference ICH Guidelines: Q6A and Q6B
3.2.S.2 製造
3.2.S.2.1 製造業者
受託者を含むすべての製造業者の名称、住所及び分担の範囲、並びに承認を得ようとする医薬品の製造及び試験に係わるすべての事業所又は施設について記載する。
3.2.S.2 Manufacture
3.2.S.2.1 Manufacturer(s)
The name, address, and responsibility of each manufacturer, including contractors, and each proposed production site or facility involved in manufacturing and testing should be provided.
3.2.S.2.2 製造方法及びプロセス・コントロール
申請者は、原薬の製造に対して、責任を持つものであり、原薬の製造方法に関して説明する必要がある。
製造方法及びプロセス・コントロールを適切に説明するため、例えば、次のような事項を記述する。
新規化学薬品:
製造工程(合成工程)の流れ図(出発物質・中間体の分子式、仕込量、収率、化学構造、試薬の分子式、仕込量、化学構造、及び原薬の分子式、収率、化学構造並びに原薬の立体化学に反映させるような事項を含む。)を示すとともに操作条件及び溶媒を明記する。
製造工程中の一連の操作手順を記述する。記述する事項は、実生産を反映する代表的なロット・スケールにおける原材料、溶媒、触媒及び試薬の量、重要工程、プロセス・コントロール、装置、操作条件(温度、圧力、pH、時間等)等である。
代替工程がある場合は、その記載も、本工程と同程度の詳細さとする。再加工を行う場合には、その工程を明らかにし、その妥当性を示す。妥当性の根拠となるデータを文献等により引用して示すか、3.2.S.2.5 において添付資料として示す。
生物薬品:
製造工程に関する情報(通例、バンク化された細胞のバイアルの使用から始まり、細胞培養、ハーベスト、精製・修飾反応、充てん、保存及び出荷条件に至る情報)を示す。
ロット及びスケール
ナンバリング・システム(未加工/未精製バルク又は中間体のプール及びロットのサイズ又はスケールに関する事項を含む。)を説明する。
細胞培養及びハーベスト
最初の播種(例えば1本以上のワーキング・セル・バンクのアンプルに含まれる細胞等を播種する)から最後のハーベスト作業に至る製造ルートのフローチャートを示す。これにはすべての工程(すなわち、種類の異なる工程単位のすべて)及び重要中間体を示し、各工程の関連事項(例えば細胞数倍加レベル、細胞濃度、容量、pH、培養時間、保持時間、温度等)を含むこと。重要工程及び規格及び試験方法を設定した重要中間体(3.2.S.2.4 参照)を明示すること。
フローチャートに示す各工程について、スケール、培地その他添加成分
(3.2.S.2.3 参照)、主な設備(詳細は3.2.A.1)及びプロセス・コントロール(工程内試験、操作管理項目、装置及び重要中間体とその規格値/判定基準(3.2.S.2.4 参照)等)を記述する。また、工程間、設備間、区画間及び建物間で材料を移送する場合には、その手順、出荷及び保存条件について記述する(保存に関する詳細は3.2.S.2.4 に記述)。
精製工程及び修飾反応
ハーベストから原薬の充てんの前までの精製工程(種類の異なる工程単位)をフローチャートで示す。
これにはすべての工程及び重要中間体並びに各工程の関連事項(例えば容量、pH、重要な操作/工程の処理時間、保持時間、温度、溶出プロファイル、画分の選定、中間体の保存等の該当する事項)を含むこと。規格及び試験方法を設定した重要工程及び重要中間体(3.2.S.2.4 参照)を明示すること。
フローチャートに示す各工程について、スケール、緩衝液その他の試薬類
(3.2.S.2.3 参照)、主な設備(詳細は3.2.A.1 )及び資材について記述する。また、工程間、設備間、区画間及び建物間で材料を移送する場合には、その手順、出荷及び保存条件について記述する(関連事項は3.2.S.2.4 に記述)。膜、クロマトグラフィー用樹脂のような資材については、使用条件及び再使用条件に関する事項を含む(設備の詳細は3.2.A.1 参照、カラム及び膜の再使用及び再生に関するバリデーション試験については3.2.S.2.5 に関連事項記載)。また、規格値/判定基準を含むプロセス・コントロール(工程内試験及び操作管理項目を含む。)を製造工程、設備及び重要中間体とともに記述する(関連事項は3.2.S.2.4 に記載)。
再加工を実施する工程について、対象とするすべての中間体又は原薬の再加工に関する基準とともに記述する(関連事項は3.2.S.2.5 に記載)。
また、工程間、設備間、区画間及び建物間で材料を移送する場合には、その手
順、出荷及び保存条件について記述する(関連事項は3.2.S.2.4 に記述)。
充てん、保存、移送(出荷)
原薬の充てん方法、プロセス・コントロール(工程内試験及び操作管理項目を含む。)及び規格/判定基準を記述する(関連事項は3.2.S.2.4 に記述)。原薬を保存する容器及び施栓系(詳細は3.2.S.6 に記述)並びに原薬の保存、移送(出荷)の条件を記述する。
参照ICHガイドライン Q5A、Q5B及びQ6B
3.2.S.2.2 Description of Manufacturing Process and Process Controls
The description of the drug substance manufacturing process represents the applicant’s commitment for the manufacture of the drug substance. Information should be provided to adequately describe the manufacturing process and process controls. For example:
NCE:
A flow diagram of the synthetic process(es) should be provided that includes molecular formulae, weights, yield ranges, chemical structures of starting materials, intermediates, reagents and drug substance reflecting stereochemistry, and identifies operating conditions and solvents.
A sequential procedural narrative of the manufacturing process should be submitted.
The narrative should include, for example, quantities of raw materials, solvents, catalysts and reagents reflecting the representative batch scale for commercial manufacture, identification of critical steps, process controls, equipment and operating conditions (e.g., temperature, pressure, pH, time).
Alternate processes should be explained and described with the same level of detail as the primary process. Reprocessing steps should be identified and justified. Any data to support this justification should be either referenced or filed in 3.2.S.2.5.
Biotech:
Information should be provided on the manufacturing process, which typically starts with a vial(s) of the cell bank, and includes cell culture, harvest(s), purification and modification reactions, filling, storage and shipping conditions.
Batch(es) and scale definition
An explanation of the batch numbering system, including information regarding any pooling of harvests or intermediates and batch size or scale should be provided.
Cell culture and harvest
A flow diagram should be provided that illustrates the manufacturing route from the original inoculum (e.g. cells contained in one or more vials(s) of the Working Cell Bank up to the last harvesting operation. The diagram should include all steps (i.e., unit operations) and intermediates. Relevant information for each stage, such as population doubling levels, cell concentration, volumes, pH, cultivation times, holding times, and temperature, should be included. Critical steps and critical intermediates for which specifications are established (as mentioned in 3.2.S.2.4) should be identified.
A description of each process step in the flow diagram should be provided. Information should be included on, for example, scale; culture media and other additives (details provided in 3.2.S.2.3); major equipment (details provided in 3.2.A.1); and process controls, including in-process tests and operational parameters, process steps, equipment and intermediates with acceptance criteria (details provided in 3.2.S.2.4).
Information on procedures used to transfer material between steps, equipment, areas, and buildings, as appropriate, and shipping and storage conditions should be provided.
(Details on shipping and storage provided in 3.2.S.2.4.)
Purification and modification reactions
A flow diagram should be provided that illustrates the purification steps (i.e., unit operations) from the crude harvest(s) up to the step preceding filling of the drug substance. All steps and intermediates and relevant information for each stage (e.g., volumes, pH, critical processing time, holding times, temperatures and elution profiles and selection of fraction, storage of intermediate, if applicable) should be included.
Critical steps for which specifications are established as mentioned in 3.2.S.2.4 should be identified.
A description of each process step (as identified in the flow diagram) should be provided. The description should include information on, for example, scale, buffers and other reagents (details provided in 3.2.S.2.3, major equipment (details provided in 3.2.A.1), and materials. For materials such as membranes and chromatography resins, information for conditions of use and reuse also should be provided. (Equipment details in 3.2.A.1; validation studies for the reuse and regeneration of columns and membranes in 3.2.S.2.5.) The description should include process controls (including inprocess tests and operational parameters) with acceptance criteria for process steps, equipment and intermediates. (Details in 3.2.S.2.4.)
Reprocessing procedures with criteria for reprocessing of any intermediate or the drug substance should be described. (Details should be given in 3.2.S.2.5.)
Information on procedures used to transfer material between steps, equipment, areas, and buildings, as appropriate, and shipping and storage conditions should be provided (details on shipping and storage provided in 3.2.S.2.4.).
Filling, storage and transportation (shipping)
A description of the filling procedure for the drug substance, process controls (including in-process tests and operational parameters), and acceptance criteria should be provided. (Details in 3.2.S.2.4.) The container closure system(s) used for storage of the drug substance (details in 3.2.S.6.) and storage and shipping conditions for the drug substance should be described.
Reference ICH Guidelines: Q5A, Q5B, and Q6B
3.2.S.2.3 原材料の管理
原薬の製造に使用される原材料(原料、出発物質、溶媒、試薬、触媒等)について、当該原材料の使用される工程を明らかにしたうえで一覧表を作成する。これらの原材料の品質及び管理について記述する。原材料(培地成分、モノクローナル抗体、酵素等の生物起源の原材料を含む。)がその使用目的に応じた規格に適合していることを裏付ける資料(外来性因子のクリアランス又は管理を含む。)
を必要に応じて示す。生物起源の原材料については、その起源、製造及び特性に関する資料を含める(詳細は3.2.A.2 に記述)。
参照ICHガイドライン Q6A及びQ6B
生物薬品:
生物起源の原材料及び出発物質の管理
生物起源の原材料に関する外来性因子の安全性評価の要約を記述する(詳細は3.2.A.2 に記述)。
細胞基材の起源、履歴及び作製
細胞基材の起源、遺伝子組換え細胞の遺伝子発現構成体の解析及びマスター・セル・バンクの作製に用いたクローン細胞について、Q5B及びQ5Dに基づいて記述する。
セル・バンクシステム、特性解析及び試験方法
セル・バンクシステム、品質管理法並びに製造及び保存中(マスター・セル・バンク及びワーキング・セル・バンクの作製手順を含む。)の細胞株の安定性について、Q5B及びQ5Dに基づいて記述する。
参照ICHガイドライン Q5A、Q5B、Q5C及びQ5D
3.2.S.2.3 Control of Materials
Materials used in the manufacture of the drug substance (e.g., raw materials, starting materials, solvents, reagents, catalysts) should be listed identifying where each material is used in the process. Information on the quality and control of these materials should be provided. Information demonstrating that materials (including biologically-sourced materials, e.g., media components, monoclonal antibodies, enzymes) meet standards appropriate for their intended use (including the clearance or control of adventitious agents) should be provided, as appropriate. For biologically-sourced materials, this can include information regarding the source, manufacture, and characterisation. (Details in 3.2.A.2 for both NCE and Biotech)
Reference ICH Guidelines: Q6A and Q6B
Biotech:
Control of Source and Starting Materials of Biological Origin
Summaries of viral safety information for biologically-sourced materials should be provided. (Details in 3.2.A.2.)
Source, history, and generation of the cell substrate
Information on the source of the cell substrate and analysis of the expression construct used to genetically modify cells and incorporated in the initial cell clone used to develop the Master Cell Bank should be provided as described in Q5B and Q5D.
Cell banking system, characterisation, and testing
Information on the cell banking system, quality control activities, and cell line stability during production and storage (including procedures used to generate the Master and Working Cell Bank(s)) should be provided as described in Q5B and Q5D.
Reference ICH Guidelines: Q5A, Q5B, Q5C and Q5D
3.2.S.2.4 重要工程及び重要中間体の管理
重要工程:製造工程のうち3.2.S.2.2 で示された重要工程において工程が管理されていることを保証するために実施される試験方法及び規格値/判定基準(その設定根拠となる試験データを含む。)を記述する。
重要中間体:製造工程中で単離される中間体の品質及び管理方法を記述する。
参照ICHガイドライン Q6A及びQ6B
生物薬品に関する追加項目:重要中間体の保存条件の妥当性を裏付ける安定性試験成績を示す。
参照ICHガイドライン Q5C
3.2.S.2.4 Controls of Critical Steps and Intermediates
Critical Steps: Tests and acceptance criteria (with justification including experimental data) performed at critical steps identified in 3.2.S.2.2 of the manufacturing process to ensure that the process is controlled should be provided.
Intermediates: Information on the quality and control of intermediates isolated during the process should be provided.
Reference ICH Guidelines: Q6A and Q6B
Additionally for Biotech: Stability data supporting storage conditions should be provided.
Reference ICH Guideline: Q5C
3.2.S.2.5 プロセス・バリデーション/プロセス評価
無菌工程及び滅菌工程のプロセス・バリデーションやプロセス評価について記述する。
生物薬品:
製造工程(再加工を行う工程を含む。)が目的に適しているかどうかを証明し、重要なプロセス・コントロール法(操作管理項目及び工程内管理試験)を選択し、重要な製造工程(細胞培養、ハーベスト、精製、修飾等)における判定基準の妥当性を実証するためのバリデーション及び評価試験に関する十分な資料を示す。試験計画並びに試験の結果、考察及び結論を記述する。試験方法とそのバリデーションについては、相互参照できるようにする(例えば3.2.S.2.4 と3.2.S.4.3 などのごとく)か、又は重要なプロセス・コントロール法の選択及び規格値/判定基準の妥当性を示す資料の一部として記述する。
ウイルス汚染を除去又は不活化する製造工程について、ウイルスクリアランス評価試験に関する資料を3.2.A.2 にて示すこと。
3.2.S.2.5 Process Validation and/or Evaluation
Process validation and/or evaluation studies for aseptic processing and sterilisation should be included.
Biotech:
Sufficient information should be provided on validation and evaluation studies to demonstrate that the manufacturing process (including reprocessing steps) is suitable for its intended purpose and to substantiate selection of critical process controls (operational parameters and in-process tests) and their limits for critical manufacturing steps (e.g., cell culture, harvesting, purification, and modification).
The plan for conducting the study should be described and the results, analysis and conclusions from the executed study(ies) should be provided. The analytical procedures and corresponding validation should be cross-referenced (e.g., 3.2.S.2.4, 3.2.S.4.3) or provided as part of justifying the selection of critical process controls and acceptance criteria.
For manufacturing steps intended to remove or inactivate viral contaminants, the information from evaluation studies should be provided in 3.2.A.2.
3.2.S.2.6 製造工程の開発の経緯
新規化学薬品:
非臨床試験や臨床試験用のロット、スケール・アップ検討時のロット、パイロット・スケール及び実生産規模(もしあれば)のロットにおいて原薬の製造工程及び製造場所に重大な変更があったときは、変更内容の説明及び考察を記述する。
3.2.S.4.4 に示された原薬のロット分析データを参照すること。
参照ICHガイドライン Q3A
生物薬品:
3.2.S.2.2 記載の製造工程の開発の経緯を記述する。承認申請資料(非臨床試験、臨床試験等)に用いられた原薬ロットにおいて製造方法の変更がなされたときは、変更内容(例:製造工程、重要な設備等)について記述し、その理由を説明すること。また、変更に関連して、開発中に製造された原薬ロットに関する事項(ロット番号、製造スケール、使途(例:安定性試験、非臨床試験、標準物質)等)を記述する。
重大な変更かどうかは、当該変更が原薬の品質(場合によっては、重要中間体の品質も含む。)にどれほど影響しうるかを評価して判断する。重大な変更と考えられるときは、当該変更の原薬の品質に対する影響の程度を判定するために、変更前後の原薬ロットを比較する試験データを提出すること(詳細はQ6Bを参照)。試験方法の選択及び試験データの妥当性を示し、結果を考察する。
製造方法の変更が原薬及びその製剤の品質に与える影響を評価するロットの比較試験には非臨床・臨床試験のロットのデータも含まれることがある。その場合には、他のモジュールに記載されているこれらの試験成績を相互参照すること。
3.2.S.4.4 に示された原薬のロット分析データを参照すること。
参照ICHガイドライン Q6B
3.2.S.2.6 Manufacturing Process Development
NCE:
A description and discussion should be provided of the significant changes made to the manufacturing process and/or manufacturing site of the drug substance used in producing nonclinical, clinical, scale-up, pilot, and, if available, production scale batches.
Reference should be made to the drug substance data provided in section 3.2.S.4.4.
Reference ICH Guideline: Q3A
Biotech:
The developmental history of the manufacturing process, as described in 3.2.S.2.2, should be provided. The description of change(s) made to the manufacture of drug substance batches used in support of the marketing application (e.g., nonclinical or clinical studies) should include, for example, changes to the process or to critical equipment. The reason for the change should be explained. Relevant information on drug substance batches manufactured during development, such as the batch number, manufacturing scale, and use (e.g., stability, nonclinical, reference material) in relation to the change, should be provided.
The significance of the change should be assessed by evaluating its potential to impact the quality of the drug substance (and/or intermediate, if appropriate). For manufacturing changes that are considered significant, data from comparative analytical testing on relevant drug substance batches should be provided to determine the impact on quality of the drug substance (see Q6B for additional guidance). A discussion of the data, including a justification for selection of the tests and assessment of results, should be included.
Testing used to assess the impact of manufacturing changes on the drug substance(s) and the corresponding drug product(s) can also include nonclinical and clinical studies.
Cross-reference to the location of these studies in other modules of the submission should be included.
Reference should be made to the drug substance data provided in section 3.2.S.4.4.
Reference ICH Guideline: Q6B
3.2.S.3 特性
3.2.S.3.1 構造その他の特性の解明
新規化学薬品:
合成経路、スペクトル分析結果等に基づいた構造決定の結果を示す。異性体存在の可能性、立体構造の決定、得られた結晶多形等についても記述する。
参照ICHガイドラインQ6A
生物薬品:
目的物質及び目的物質関連物質について、適宜、一次構造、二次構造及び高次構造、翻訳後の構造(糖結合形等)、生物活性、純度並びに免疫化学的性質を明らかにすること。
参照ICHガイドラインQ6B
3.2.S.3.2 不純物
不純物について記述する。
参照ICHガイドラインQ3A、Q3C、Q5C、Q6A及びQ6B
3.2.S.3 Characterisation
3.2.S.3.1 Elucidation of Structure and other Characteristics
NCE:
Confirmation of structure based on e.g., synthetic route and spectral analyses should be provided. Information such as the potential for isomerism, the identification of stereochemistry, or the potential for forming polymorphs should also be included.
Reference ICH Guideline: Q6A
Biotech:
For desired product and product-related substances, details should be provided on primary, secondary and higher-order structure, post-translational forms (e.g., glycoforms), biological activity, purity, and immunochemical properties, when relevant.
Reference ICH Guideline: Q6B
3.2.S.3.2 Impurities
Information on impurities should be provided.
Reference ICH Guidelines: Q3A, Q3C, Q5C, Q6A, and Q6B
3.2.S.4 原薬の管理
3.2.S.4.1 規格及び試験方法
原薬の規格及び試験方法を示す。
参照ICHガイドライン Q6A及びQ6B
3.2.S.4.2 試験方法(分析方法)
原薬の規格及び試験方法における試験方法の詳細を示す。
参照ICHガイドライン Q2A及びQ6B
3.2.S.4.3 試験方法(分析方法)のバリデーション
原薬の試験方法の分析法バリデーションについて、試験成績を示し、記述する。
参照ICHガイドライン Q2A、Q2B及びQ6B
3.2.S.4.4 ロット分析
ロット及びロット分析結果について記述する。
参照ICHガイドライン Q3A、Q3C、Q6A及びQ6B
3.2.S.4.5 規格及び試験方法の妥当性
原薬の規格及び試験方法の妥当性について記述する。
参照ICHガイドライン Q3A、Q3C、Q6A及びQ6B
3.2.S.4 Control of Drug Substance
3.2.S.4.1 Specification
The specification for the drug substance should be provided.
Reference ICH Guidelines: Q6A and Q6B
3.2.S.4.2 Analytical Procedures
The analytical procedures used for testing the drug substance should be provided.
Reference ICH Guidelines: Q2A and Q6B
3.2.S.4.3 Validation of Analytical Procedures
Analytical validation information, including experimental data for the analytical procedures used for testing the drug substance, should be provided.
Reference ICH Guidelines: Q2A, Q2B, and Q6B
3.2.S.4.4 Batch Analyses
Description of batches and results of batch analyses should be provided.
Reference ICH Guidelines: Q3A, Q3C, Q6A, and Q6B
3.2.S.4.5 Justification of Specification
Justification for the drug substance specification should be provided.
Reference ICH Guidelines: Q3A, Q3C, Q6A and Q6B
3.2.S.5 標準品又は標準物質
原薬の試験に用いられる標準品又は標準物質について記述する。
参照ICHガイドラインQ6A及びQ6B
3.2.S.5 Reference Standards or Materials
Information on the reference standards or reference materials used for testing of the drug substance should be provided.
Reference ICH Guidelines: Q6A and Q6Bs
3.2.S 原薬
複数の原薬を含む製剤にあっては、各原薬ごとに3.2.S項の資料を作成する。
3.2.S Drug Substance
For a drug product containing more than one drug substance, the information requested for part “S” should be provided in its entirety for each drug substance.
3.2.S.6 容器及び施栓系
容器及び施栓系について、一次包装を構成する各素材を明らかにすることを含め、記述する。また、容器及び施栓系の規格及び試験方法を記述する。
規格及び試験方法には外観・性状及び確認試験(その他、重要と思われるものについては、適宜、寸法を図示すること)が含まれる。必要に応じて、公定書にない試験方法を含め、そのバリデーションとともに示すこと。
機能を有しない二次包装材(例えば、追加保護機能のないもの等)については、外観・形状に関する簡潔な記述のみでよい。機能を有する二次包装材については、追加される機能に関して記述する。
容器及び施栓系の適格性について、素材の選択、防湿性、遮光性、素材と原薬との適合性等の観点から、容器への吸着、溶出や素材の安全性を示すことを含めて記述する。
3.2.S.6 Container Closure System
A description of the container closure system(s) should be provided, including the identity of materials of construction of each primary packaging component, and their specifications.
The specifications should include description and identification (and critical dimensions with drawings, where appropriate). Non-compendial methods (with validation) should be included, where appropriate.
For non-functional secondary packaging components (e.g., those that do not provide additional protection), only a brief description should be provided. For functional secondary packaging components, additional information should be provided.
The suitability should be discussed with respect to, for example, choice of materials, protection from moisture and light, compatibility of the materials of construction with the drug substance, including sorption to container and leaching, and/or safety of materials of construction.
3.2.S.7 安定性
3.2.S.7.1 安定性のまとめ及び結論
実施された試験の種類、試験計画及び試験結果の要約を示す。苛酷試験、加速試験等の結果を含める。保存条件に関する結論及び必要に応じてリテスト期間又は有効期間に関する結論をまとめる。
参照ICHガイドラインQ1A、Q1B及びQ5C
3.2.S.7.2 承認後の安定性試験計画の作成及び実施
承認後の安定性試験計画及び試験データの取扱い方を明らかにしておくこと。
参照ICHガイドラインQ1A及びQ5C
3.2.S.7.3 安定性データ
表、グラフ、文章等適切な方法で安定性試験(苛酷試験、加速試験等)の結果を示すこと。分析方法及びそのバリデーションについても記述する。
参照ICHガイドラインQ1A、Q1B、Q2A、Q2B及びQ5C
3.2.S.7 Stability
3.2.S.7.1 Stability Summary and Conclusions
The types of studies conducted, protocols used, and the results of the studies should be summarized. The summary should include results, for example, from forced degradation studies and stress conditions, as well as conclusions with respect to storage conditions and retest date or shelf-life, as appropriate.
Reference ICH Guidelines: Q1A, Q1B, and Q5C
3.2.S.7.2 Post-approval Stability Protocol and Stability Commitment
The post-approval stability protocol and stability commitment should be provided.
Reference ICH Guidelines: Q1A and Q5C
3.2.S.7.3 Stability Data
Results of the stability studies (e.g., forced degradation studies and stress conditions) should be presented in an appropriate format such as tabular, graphical, or narrative. Information on the analytical procedures used to generate the data and validation of these procedures should be included.
Reference ICH Guidelines: Q1A, Q1B, Q2A, Q2B, and Q5C
3.2.P 製剤
3.2.P.1 製剤及び処方
製剤及びその処方について記述する。記述する事項の例は次のとおりである。
- 剤型
- 成分分量、すなわち、剤型中の全成分の一覧、単位当たりの分量(過量仕込みがあれば、それを含む。)、配合目的及び準拠すべき品質規格/基準(公定書各条によるのか自社規格及び試験方法等によるのか)を記述する。
- 添付溶解液
- 製剤及び添付溶解液に使用する容器及び施栓系の種類を適宜、記述する。
参照ICHガイドラインQ6A及びQ6B
3.2.P Drug Product
3.2.P.1 Description and Composition of the Drug Product
A description of the drug product and its composition should be provided. The information provided should include, for example:
- Description* of the dosage form;
- Composition, i.e., list of all components of the dosage form, and their amount on a per unit basis (including overages, if any) the function of the components, and a reference to their quality standards (e.g., compendial monographs or manufacturer’s specifications)
- Description of accompanying reconstitution diluent(s); and
- Type of container and closure used for the dosage form and accompanying reconstitution diluent, if applicable.
Reference ICH Guidelines: Q6A and Q6B
* For a drug product supplied with reconstitution diluent(s), the information on the diluent(s) should be provided in a separate part “P”, as appropriate
3.2.P.2 製剤開発の経緯
製剤開発の経緯の項には、剤型、製剤設計・処方、製造工程、容器及び施栓系、微生物学的観点から見た特徴及び使用方法等が、使用目的に叶うことを裏付けるために実施された開発段階での検討について記述する。本項に記述する試験は、規格及び試験方法に基づいて実施する品質管理のためのルーチン試験とは区別すべきものである。更に、再現性のあるロット生産、製剤機能、製剤の品質に影響すると考えられる製剤設計、処方、製剤化工程の特徴的指標(重要なパラメータ)について明らかにし、説明すること。個別に実施した試験又は文献から得られた裏付けデータや結果は本項に含めるか、別添とする。(製剤機能に関する)追加データについては、申請資料の非臨床又は臨床の項を参照してもよい。
参照ICHガイドラインQ6A及びQ6B
3.2.P.2 Pharmaceutical Development
The Pharmaceutical Development section should contain information on the development studies conducted to establish that the dosage form, the formulation, manufacturing process, container closure system, microbiological attributes and usage instructions are appropriate for the purpose specified in the application. The studies described here are distinguished from routine control tests conducted according to specifications. Additionally, this section should identify and describe the formulation and process attributes (critical parameters) that can influence batch reproducibility, product performance and drug product quality. Supportive data and results from specific studies or published literature can be included within or attached to the Pharmaceutical Development section. Additional supportive data can be referenced to the relevant nonclinical or clinical sections of the application.
Reference ICH Guidelines: Q6A and Q6B
3.2.P.2.1 製剤成分
3.2.P.2.1.1 原薬
原薬と3.2.P.1 に記載の添加剤との配合適性を考察する。製剤機能に影響する可能性がある原薬の重要な物理的化学的性質(水分含量、溶解性、粒度分布、結晶多形、固体状態での存在形等)を記載し、考察する。複数の原薬を含む製剤(配合剤等)については、原薬相互の配合適性を考察する。
3.2.P.2.1 Components of the Drug Product
3.2.P.2.1.1 Drug Substance
The compatibility of the drug substance with excipients listed in 3.2.P.1 should be discussed. Additionally, key physicochemical characteristics (e.g., water content, solubility, particle size distribution, polymorphic or solid state form) of the drug substance that can influence the performance of the drug product should be discussed. For combination products, the compatibility of drug substances with each other should be discussed.
3.2.P.2.1.2 添加剤
3.2.P.1 に記載の添加剤について、その選択理由、添加量及び製剤機能に影響する可能性がある特性を各添加剤の機能と関連づけて考察する。
3.2.P.2.1.2 Excipients
The choice of excipients listed in 3.2.P.1, their concentration, their characteristics that can influence the drug product performance should be discussed relative to their respective functions.
3.2.P.2.2 製剤
3.2.P.2.2.1 製剤設計
申請する投与経路及び用法を考慮して、製剤設計の簡潔な要約を示す。臨床試験に用いられた製剤処方と3.2.P.1 に記述した製剤処方とが異なるときは、その違いについて考察する。必要に応じ、製剤の同等・同質性に係わるin vitro試験(溶出試験等)又はin vivo 試験(生物学的同等性試験等)の試験結果について考察する。
3.2.P.2.2 Drug Product
3.2.P.2.2.1 Formulation Development
A brief summary describing the development of the drug product should be provided, taking into consideration the proposed route of administration and usage. The differences between clinical formulations and the formulation (i.e. composition) described in 3.2.P.1 should be discussed. Results from comparative in vitro studies (e.g., dissolution) or comparative in vivo studies (e.g., bioequivalence) should be discussed when appropriate.
3.2.P.2.2.2 過量仕込み
3.2.P.1 に製剤処方の過量仕込みが記載されているときは、その妥当性を示す。
3.2.P.2.2.3 物理的化学的及び生物学的性質
製剤特性に関係した事項(例:pH、イオン強度、溶出特性、分散性、再調製の際の溶解性、粒度分布、凝集性、結晶多形、レオロジー特性、生物活性/力価、免疫学的性質等)について記述する。
3.2.P.2.2.2 Overages
Any overages in the formulation(s) described in 3.2.P.1 should be justified.
3.2.P.2.2.3 Physicochemical and Biological Properties
Parameters relevant to the performance of the drug product, such as pH, ionic strength, dissolution, redispersion, reconstitution, particle size distribution, aggregation, polymorphism, rheological properties, biological activity or potency, and/or immunological activity, should be addressed.
3.2.P.2.3 製造工程の開発の経緯
3.2.P.3.3 記載の製造工程の選択及び最適化について、特に重要な点を説明する。適宜、滅菌方法について説明し、その妥当性を示す。主要な臨床試験に用いたロットの製造工程と3.2.P.3.3 記載の製造工程との違いが製剤特性に影響を与えうるときは、それについて考察する。
3.2.P.2.3 Manufacturing Process Development
The selection and optimisation of the manufacturing process described in 3.2.P.3.3, in particular its critical aspects, should be explained. Where relevant, the method of sterilisation should be explained and justified. Differences between the manufacturing process(es) used to produce pivotal clinical batches and the process described in 3.2.P.3.3 that can influence the performance of the product should be discussed.
3.2.P.2.4 容器及び施栓系
製剤の保存、移送(出荷)及び使用時に用いられる容器及び施栓系の適格性(3.2.P.7 参照)について考察する。これには、素材の選択、防湿性・遮光性、構成する素材と製剤との適合性(容器への吸着・溶出を含む)、構成する素材の安全性、性能(製剤の一部として申請されている場合は容器/用具からの注出量の再現性等)等がある。
3.2.P.2.4 Container Closure System
The suitability of the container closure system (described in 3.2.P.7) used for the storage, transportation (shipping) and use of the drug product should be discussed. This discussion should consider, e.g., choice of materials, protection from moisture and light, compatibility of the materials of construction with the dosage form (including sorption to container and leaching) safety of materials of construction, and performance (such as reproducibility of the dose delivery from the device when presented as part of the drug product).
3.2.P.2.5 微生物学的観点からみた特徴
必要に応じて、製剤の微生物学的観点からみた特徴(例えば非無菌製剤の微生物限度試験を行わないことの根拠、抗菌効果のある保存剤を含有する製剤にあっては、その選択理由及び効力を含む。)について考察する。無菌製剤の場合、微生物汚染を防ぐための容器及び施栓系の完全性を示す。
3.2.P.2.5 Microbiological Attributes
Where appropriate, the microbiological attributes of the dosage form should be discussed, including, for example, the rationale for not performing microbial limits testing for nonsterile products and the selection and effectiveness of preservative systems in products containing antimicrobial preservatives. For sterile products, the integrity of the container closure system to prevent microbial contamination should be addressed.
3.2.P.2.6 溶解液や使用時の容器/用具との適合性
製剤と溶解液や使用時の容器/用具との適合性(溶液中の原薬の沈殿、注射用容器への吸着、安定性等)について記述し、適切かつ必要な情報が添付文書等に記載できるようにする。
3.2.P.2.6 Compatibility
The compatibility of the drug product with reconstitution diluent(s) or dosage devices (e.g., precipitation of drug substance in solution, sorption on injection vessels, stability) should be addressed to provide appropriate and supportive information for the labeling.
3.2.P.3 製造
3.2.P.3.1 製造者
受託者を含むすべての製造業者の名称、住所及び分担の範囲、並びに承認を得ようとする医薬品の製造及び試験に係わるすべての事業所又は施設について記載する。
3.2.P.3 Manufacture
3.2.P.3.1 Manufacturer(s)
The name, address, and responsibility of each manufacturer, including contractors, and each proposed production site or facility involved in manufacturing and testing should be provided.
3.2.P.3.2 製造処方
製剤の製造工程に使用するすべての成分の一覧、ロット当たりの分量(過量仕込みがあれば、それを含む。)及び準拠すべき品質規格/基準を記載する。
3.2.P.3.2 Batch Formula
A batch formula should be provided that includes a list of all components of the dosage form to be used in the manufacturing process, their amounts on a per batch basis, including overages, and a reference to their quality standards.
3.2.P.3.3 製造工程及びプロセス・コントロール
製造工程の各工程及び各材料がどの工程で入ってくるかを示した流れ図を記載する。プロセス・コントロール、中間体試験又は最終的な製品管理が実施される重要工程及び重要点を明示する。包装工程を含む製造工程について、各工程の順序及び製造規模を記述する。製剤の品質に直接影響する新規の工程または技術及び包装作業について特に詳細に記述する。製造設備について、少なくとも、関連する機器の種類(タンブル・ブレンダー、インライン・ホモゲナイザー等)及び製造能力を示す。製造工程の各工程について、時間、温度、pH等適切なプロセス・パラメータを示す。パラメータの数値は、目標としたい範囲で示すことができる。重要工程に関するパラメータの目標としたい数値範囲については、3.2.P.3.4 でその妥当性を説明すること。環境条件(発泡製剤のための低湿度条件等)についての記載が必要な場合もある。
製品の再加工を提案する場合は、その妥当性を説明しなければならない。妥当性の根拠資料は、本項に資料又は参考資料として示す。
生物薬品について、適宜、3.2.A.1(製造施設及び設備)を参照する。
参照ICHガイドラインQ6B
3.2.P.3.3 Description of Manufacturing Process and Process Controls
A flow diagram should be presented giving the steps of the process and showing where materials enter the process. The critical steps and points at which process controls, intermediate tests or final product controls are conducted should be identified. A narrative description of the manufacturing process, including packaging, that represents the sequence of steps undertaken and the scale of production should also be provided. Novel processes or technologies and packaging operations that directly affect product quality should be described with a greater level of detail. Equipment should, at least, be identified by type (e.g., tumble blender, in-line homogeniser) and working capacity, where relevant. Steps in the process should have the appropriate process parameters identified, such as time, temperature, or pH. Associated numeric values can be presented as an expected range. Numeric ranges for critical steps should be justified in Section 3.2.P.3.4. In certain cases, environmental conditions (e.g., low humidity for an effervescent product) should be stated.
Proposals for the reprocessing of materials should be justified. Any data to support this justification should be either referenced or filed in this section (3.2.P.3.3).
Additionally for Biotech see 3.2.A.1 for facilities, if appropriate.
Reference ICH Guideline: Q6B
3.2.P.3.4 重要工程及び重要中間体の管理
重要工程:製造工程のうち3.2.P.3.3 で示された重要工程において工程が適切に管理されていることを保証するために実施される試験方法/判定基準(その設定根拠となる試験データを含む。)を記述する。
重要中間体:製造工程中で単離される中間体の品質及び管理方法を記述する。
参照ICHガイドラインQ2A、Q2B、Q6A及びQ6B
3.2.P.3.4 Controls of Critical Steps and Intermediates
Critical Steps: Tests and acceptance criteria should be provided (with justification, including experimental data) performed at the critical steps identified in 3.2.P.3.3 of the manufacturing process, to ensure that the process is controlled.
Intermediates: Information on the quality and control of intermediates isolated during the process should be provided.
Reference ICH Guidelines: Q2A, Q2B, Q6A, and Q6B
3.2.P.3.5 プロセス・バリデーション/プロセス評価
製造工程における重要工程や重要試験に関するプロセス・バリデーション/プロセス評価(滅菌工程、無菌工程又は充てん工程のバリデーション等)の記述、文書化及び結果について記載する。必要に応じ、ウイルス安全性評価について、3.2.A.2 に記載する。
参照ICHガイドラインQ6B
3.2.P.3.5 Process Validation and/or Evaluation
Description, documentation, and results of the validation and/or evaluation studies should be provided for critical steps or critical assays used in the manufacturing process (e.g., validation of the sterilisation process or aseptic processing or filling). Viral safety evaluation should be provided in 3.2.A.2, if necessary.
Reference ICH Guideline: Q6B
3.2.P.4 添加剤の管理
3.2.P.4.1 規格及び試験方法
添加剤の規格及び試験方法を示す。
参照ICHガイドラインQ6A及びQ6B
3.2.P.4.2 試験方法(分析方法)
適宜、添加剤の規格及び試験方法における試験方法の詳細を示す。
参照ICHガイドラインQ2A及びQ6B
3.2.P.4.3 試験方法(分析方法)のバリデーション
添加剤の試験方法の分析法バリデーションについて、試験成績を示し、記述する。
参照ICHガイドラインQ2A、Q2B及びQ6B
3.2.P.4.4 規格及び試験方法の妥当性
適宜、添加剤の規格設定の妥当性について記述する。
参照ICHガイドラインQ3C及びQ6B
3.2.P.4.5 ヒト又は動物起源の添加剤
ヒト又は動物起源の添加剤について、外来性因子に関する情報(起原、規格及び試験方法、実施された試験に関する記述、ウイルス安全性データ等)を示す(詳細は3.2.A.2に記述)。
参照ICHガイドラインQ5A、Q5D及びQ6B
3.2.P.4.6 新規添加剤
製剤に初めて使用される添加剤又は新投与経路で使用される添加剤について、安全性データ(非臨床/臨床)を参照しつつ、製造方法、特性及び品質管理法を原薬と同様、記述する(詳細は3.2.A.3 に記述)。
3.2.P.4 Control of Excipients
3.2.P.4.1 Specifications
The specifications for excipients should be provided.
Reference ICH Guideline: Q6A and Q6B
3.2.P.4.2 Analytical Procedures
The analytical procedures used for testing the excipients should be provided, where appropriate.
Reference ICH Guidelines: Q2A and Q6B
3.2.P.4.3 Validation of Analytical Procedures
Analytical validation information, including experimental data, for the analytical procedures used for testing the excipients should be provided, where appropriate.
Reference ICH Guidelines: Q2A, Q2B, and Q6B
3.2.P.4.4 Justification of Specifications
Justification for the proposed excipient specifications should be provided, where appropriate.
Reference ICH Guidelines: Q3C and Q6B
3.2.P.4.5 Excipients of Human or Animal Origin
For excipients of human or animal origin, information should be provided regarding adventitious agents (e.g., sources, specifications; description of the testing performed; viral safety data). (Details in 3.2.A.2).
Reference ICH Guidelines: Q5A, Q5D, and Q6B
3.2.P.4.6 Novel Excipients
For excipient(s) used for the first time in a drug product or by a new route of administration, full details of manufacture, characterisation, and controls, with cross references to supporting safety data (nonclinical and/or clinical) should be provided according to the drug substance format. (Details in 3.2.A.3).
3.2.P.5 製剤の管理
3.2.P.5.1 規格及び試験方法
製剤の規格及び試験方法を示す。
参照ICHガイドラインQ3B、Q6A及びQ6B
3.2.P.5.2 試験方法(分析方法)
製剤の規格及び試験方法における試験方法の詳細を示す。
参照ICHガイドラインQ2A及びQ6B
3.2.P.5.3 試験方法(分析方法)のバリデーション
製剤の試験方法の分析法バリデーションについて、試験成績を示し、記述する。
参照ICHガイドラインQ2A、Q2B及びQ6B
3.2.P.5.4 ロット分析
ロット及びロット分析結果について記述する。
参照ICHガイドラインQ3B、Q3C、Q6A及びQ6B
3.2.P.5.5 不純物の特性
3.2.S.3.2(不純物)の項に記載していない不純物については、その特性に関する情報を記述する。
参照ICHガイドラインQ3B、Q5C、Q6A及びQ6B
3.2.P.5.6 規格及び試験方法の妥当性
製剤の規格及び試験方法の妥当性について記載する。
参照ICHガイドラインQ3B、Q6A及びQ6B
3.2.P.5 Control of Drug Product
3.2.P.5.1 Specification(s)
The specification(s) for the drug product should be provided.
Reference ICH Guidelines: Q3B, Q6A and Q6B
3.2.P.5.2 Analytical Procedures
The analytical procedures used for testing the drug product should be provided.
Reference ICH Guidelines: Q2A and Q6B
3.2.P.5.3 Validation of Analytical Procedures
Analytical validation information, including experimental data, for the analytical procedures used for testing the drug product, should be provided.
Reference ICH Guidelines: Q2A, Q2B and Q6B
3.2.P.5.4 Batch Analyses
A description of batches and results of batch analyses should be provided.
Reference ICH Guidelines: Q3B, Q3C, Q6A, and Q6B
3.2.P.5.5 Characterisation of Impurities
Information on the characterisation of impurities should be provided, if not previously provided in “3.2.S.3.2 Impurities”.
Reference ICH Guidelines: Q3B, Q5C, Q6A, and Q6B
3.2.P.5.6 Justification of Specification(s)
Justification for the proposed drug product specification(s) should be provided.
Reference ICH Guidelines: Q3B, Q6A, and Q6B
3.2.P.6 標準品又は標準物質
3.2.S.5 の項に記載していない標準品又は標準物質を製剤の試験に用いる場合には、それらに関する情報を記載する。
参照ICHガイドラインQ6A及びQ6B
3.2.P.6 Reference Standards or Materials
Information on the reference standards or reference materials used for testing of the drug product should be provided, if not previously provided in “3.2.S.5 Reference Standards or Materials”.
Reference ICH Guidelines: Q6A and Q6B
3.2.P.7 容器及び施栓系
容器及び施栓系について、一次包装を構成する各素材を明らかにすることを含め、記述する。また、容器及び施栓系の規格及び試験方法を記述する。規格及び試験方法には外観・性状及び確認試験(その他、重要と思われるものについては、適宜、寸法を図示すること)が含まれる。試験方法については、必要に応じて、公定書にない試験方法を含め、そのバリデーションとともに示すこと。
機能を有しない二次包装材(例えば、追加保護機能のないもの、製剤の輸送に関与しないもの等)については、外観・形状に関する簡潔な記述のみでよい。機能を有する二次包装材については、追加される機能に関して記述する。
容器及び施栓系の適格性については3.2.P.2 に記述する。
3.2.P.7 Container Closure System
A description of the container closure systems should be provided, including the identity of materials of construction of each primary packaging component and its specification. The specifications should include description and identification (and critical dimensions, with drawings where appropriate). Non-compendial methods (with validation) should be included where appropriate.
For non-functional secondary packaging components (e.g., those that neither provide additional protection nor serve to deliver the product), only a brief description should be provided. For functional secondary packaging components, additional information should be provided.
Suitability information should be located in 3.2.P.2.
3.2.P.8 安定性
3.2.P.8.1 安定性のまとめ及び結論
実施された試験の種類、試験計画及び試験結果の要約を示す。要約には、例えば、保存条件及び有効期間を含める。また、適宜、使用時の保存条件及び有効期間に関する結論をまとめる。
参照ICHガイドラインQ1A、Q1B、Q3B、Q5C及びQ6A
3.2.P.8.2 承認後の安定性試験計画の作成及び実施
承認後の安定性試験計画及び試験データの取扱い方を明らかにしておくこと。
参照ICHガイドラインQ1A及びQ5C
3.2.P.8.3 安定性データ
表、グラフ、文章等適切な方法で安定性試験結果を示すこと。試験方法及びそのバリデーションについても記述する。
不純物の特性は、3.2.P.5.5 に記載する。
参照ICHガイドラインQ1A、Q1B、Q2A、Q2B及びQ5C
3.2.P.8 Stability
3.2.P.8.1 Stability Summary and Conclusion
The types of studies conducted, protocols used, and the results of the studies should be summarized. The summary should include, for example, conclusions with respect to storage conditions and shelf-life, and, if applicable, in-use storage conditions and shelf-life.
Reference ICH Guidelines: Q1A, Q1B, Q3B, and Q5C, Q6A
3.2.P.8.2 Post-approval Stability Protocol and Stability Commitment
The post-approval stability protocol and stability commitment should be provided.
Reference ICH Guidelines: Q1A and Q5C
3.2.P.8.3 Stability Data
Results of the stability studies should be presented in an appropriate format (e.g. tabular, graphical, narrative). Information on the analytical procedures used to generate the data and validation of these procedures should be included.
Information on characterisation of impurities is located in 3.2.P.5.5.
Reference ICH Guidelines: Q1A, Q1B, Q2A, Q2B and Q5C
3.2.A その他
3.2.A.1 製造施設及び設備
生物薬品:
原材料、作業従事者、廃棄物及び中間体の製造区域への出入りを含む流れ図を示す。医薬品(原薬及び製剤をいう。以下同じ。)の品質確保に関連すると思われる隣接区域又は部屋について記述する。
申請に係る医薬品と同一区域で製造され、又は取り扱われたすべての開発中又は既承認の医薬品について記述する。
医薬品と接触する装置及びその使用法(専用又は共用)について概略を記述する。適宜、各器具・材料の調製、洗浄、滅菌及び保管方法について概略を記述する。
細胞バンク調製・医薬品製造について、操作手順(例:洗浄、製造スケジュール等)及び区域・設備の汚染・交叉汚染防止のための設計仕様(例:区域の等級区分等)を記述する。
3.2.A APPENDICES
3.2.A.1 Facilities and Equipment
Biotech:
A diagram should be provided illustrating the manufacturing flow including movement of raw materials, personnel, waste, and intermediate(s) in and out of the manufacturing areas. Information should be presented with respect to adjacent areas or rooms that may be of concern for maintaining integrity of the product.
Information on all developmental or approved products manufactured or manipulated in the same areas as the applicant’s product should be included.
A summary description of product-contact equipment, and its use (dedicated or multiuse) should be provided. Information on preparation, cleaning, sterilisation, and storage of specified equipment and materials should be included, as appropriate.
Information should be included on procedures (e.g., cleaning and production scheduling) and design features of the facility (e.g., area classifications) to prevent contamination or cross-contamination of areas and equipment, where operations for the preparation of cell banks and product manufacturing are performed.
3.2.A.2 外来性感染性物質の安全性評価
外来性感染性物質による汚染の可能性について安全性を評価する資料を示す。
非ウイルス性感染性物質:
非ウイルス性感染性物質(伝達性海綿状脳症関連物質、細菌、マイコプラズマ、真菌等)について、その混入防止及びコントロール法を記述する。これには、例えば、原材料・添加剤に関する証明資料や試験結果などが含まれる。これは、各原材料や添加剤、プロセス及び感染性物質それぞれについて適切な情報であればよい。
参照ICHガイドラインQ5A、Q5D及びQ6B
外来性ウイルス:
ウイルス安全性評価試験について本項に詳細に記述する。ウイルス安全性評価試験では、製造に使用する原材料等が安全であり、かつ、製造中に起こりうる汚染の可能性を試験、評価、あるいは排除する方策が適切であることを示す必要がある。
参照ICHガイドラインQ5A、Q5D及びQ6B
生物起源の原材料
動物又はヒト起源の原材料(体液、組織、臓器、細胞株等)のウイルス安全性評価に必要な事項を記述する(関連事項について3.2.S.2.3 及び3.2.P.4.5 参照)。細胞株に関してウイルス安全性の観点からの細胞の選択、試験及び評価並びにセル・バンクに関してウイルス安全性上の適格性について記述する(関連事項について3.2.S.2.3 参照)。
製造工程の適切な段階における試験
製造工程(細胞基材、未加工/未精製バルク又はウイルスクリアランス試験後の段階等)において行うウイルス試験の選択について根拠を示す。可能な範囲で、試験の種類、感度、特異性、実施頻度についても記述する。製造工程の適切な段階において行う試験によって、医薬品がウイルスに汚染されていないことを確認した結果を示す(関連事項について3.2.S.2.4 及び3.2.P.3.4 参照)。
未加工/未精製バルクのウイルス試験
Q5A及びQ6Bに従って実施した未加工/未精製バルクのウイルス試験の結果を記述する。
ウイルスクリアランス試験
Q5Aに従ってウイルスクリアランス評価試験を行う際の考え方と実施要領を試験結果及び評価とともに記述する。実生産スケールの製造プロセスと比較したスケール・ダウン・モデルの妥当性、製造設備・資材に対するウイルス不活化・除去方法の妥当性、並びに製造工程がウイルス不活化・除去能力を有することを示す資料を含むこと(関連事項について3.2.S.2.5 及び3.2.P.3.5 参照)。
参照ICHガイドラインQ5A、Q5D及びQ6B
3.2.A.2 Adventitious Agents Safety Evaluation
Information assessing the risk with respect to potential contamination with adventitious agents should be provided in this section.
For non-viral adventitious agents:
Detailed information should be provided on the avoidance and control of non-viral adventitious agents (e.g., transmissible spongiform encephalopathy agents, bacteria, mycoplasma, fungi). This information can include, for example, certification and/or testing of raw materials and excipients, and control of the production process, as appropriate for the material, process and agent.
Reference ICH Guidelines: Q5A, Q5D, and Q6B
For viral adventitious agents:
Detailed information from viral safety evaluation studies should be provided in this section.
Viral evaluation studies should demonstrate that the materials used in production are considered safe, and that the approaches used to test, evaluate, and eliminate the potential risks during manufacturing are suitable.
The applicant should refer to Q5A, Q5D, and Q6B for further guidance.
Materials of Biological Origin
Information essential to evaluate the virological safety of materials of animal or human origin (e.g. biological fluids, tissue, organ, cell lines) should be provided. (See related information in 3.2.S.2.3, and 3.2.P.4.5). For cell lines, information on the selection, testing, and safety assessment for potential viral contamination of the cells and viral qualification of cell banks should also be provided. (See related information in 3.2.S.2.3).
Testing at appropriate stages of production
The selection of virological tests that are conducted during manufacturing (e.g., cell substrate, unprocessed bulk or post viral clearance testing) should be justified. The type of test, sensitivity and specificity of the test, if applicable, and frequency of testing should be included. Test results to confirm, at an appropriate stage of manufacture, that the product is free from viral contamination should be provided. (See related information in 3.2.S.2.4 and 3.2.P.3.4).
Viral Testing of Unprocessed Bulk
In accordance with Q5A and Q6B, results for viral testing of unprocessed bulk should be included.
Viral Clearance Studies
In accordance with Q5A, the rationale and action plan for assessing viral clearance and the results and evaluation of the viral clearance studies should be provided. Data can include those that demonstrate the validity of the scaled-down model compared to the commercial scale process; the adequacy of viral inactivation or removal procedures for manufacturing equipment and materials; and manufacturing steps that are capable of removing or inactivating viruses. (See related information in 3.2.S.2.5 and 3.2.P.3.5).
Reference ICH Guidelines: Q5A, Q5D, and Q6B
3.2.A.3 新規添加剤
3.2.A.3 Excipients
3.2.R 各極の要求資料
原薬及び製剤に関して各極に特有な追加要求資料は3.2.R 項として提出する。
詳細については、各極の関連ガイドラインを参照するか規制当局に相談すること。
本件に関係する項目としては以下のような例がある。
- バッチ・レコード(米国)
- メソッド・バリデーション・パッケージ(米国)
- コンパラビリティー・プロトコール(米国)
- 製剤のプロセス・バリデーション・スキーム(EU)
バリデーションが完了していないときは、実施しようとする試験の要約を示す。
- 医療用具(EU)
3.2.R Regional Information
Any additional drug substance and/or drug product information specific to each region should be provided in section R of the application.
Applicants should consult the appropriate regional guidelines and/or regulatory authorities for additional guidance.
Some examples are as follows:
- Executed Batch Records (USA only)
- Method Validation Package (USA only)
- Comparability Protocols (USA only)
- Process Validation Scheme for the Drug Product (EU only)
Where validation is still to be completed, a summary of the studies intended to be conducted should be provided.
- Medical Device (EU only)
3.3 参考文献
適宜、参考文献を示す。
3.3 Literature References
Key literature referenced should be provided, if applicable.





