

医薬審発第899号 CTD通知(別紙 3)
CTD-品質に関する文書の作成要領に関するガイドライン
(2002年9月11-12日ワシントン会議修正版)
ICH HARMONISED GUIDELINE
QUALITY – M4Q –
(Numbering and Section Headers have been edited for consistency and use in e-CTD as agreed at the Washington DC Meeting, September 11-12, 2002)
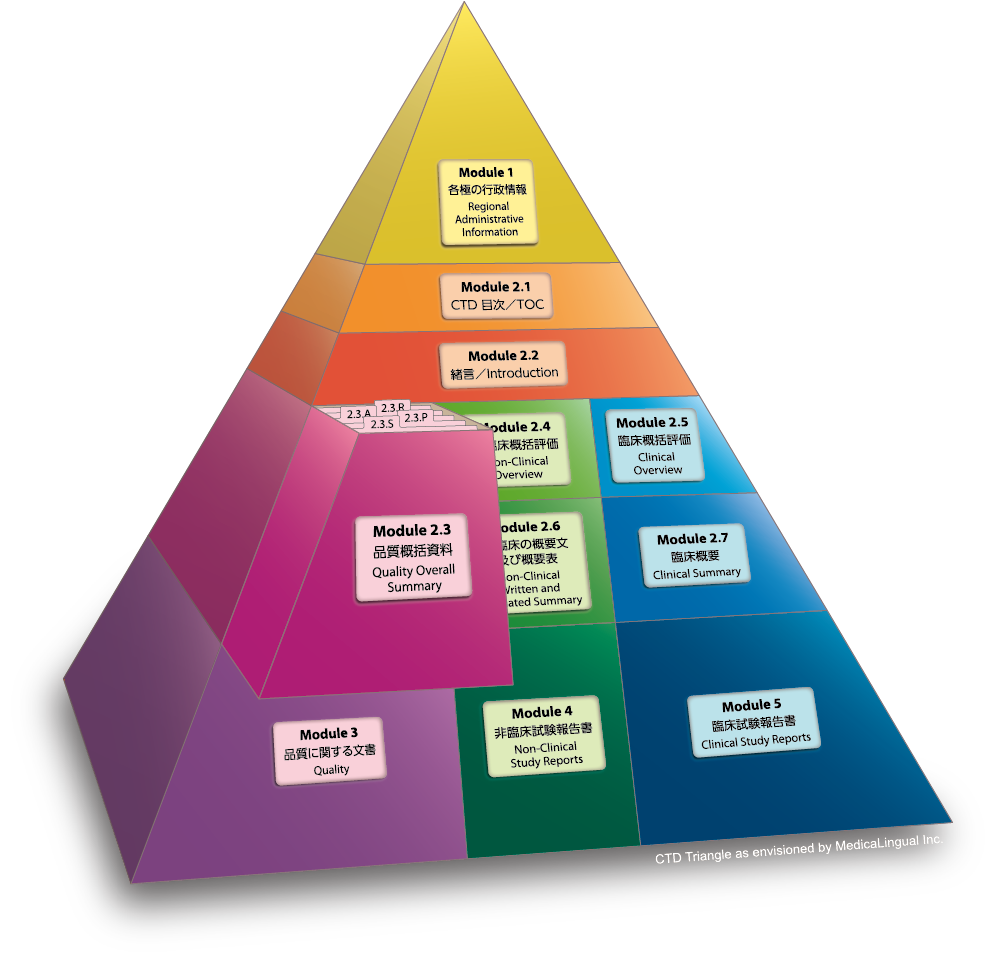
2.3 品質に関する概括資料
品質に関する概括資料(以下「QOS」という。)は、第3部の資料をその範囲及び構成に即して要約したものである。QOSには、第3部(モジュール3)及びCTDの他の部分に含まれていない事項、データ及び考察を加えてはならない。
2.3 QUALITY OVERALL SUMMARY
The Quality Overall Summary (QOS) is a summary that follows the scope and the outline of the Body of Data in Module 3. The QOS should not include information, data or justification that was not already included in Module 3 or in other parts of the CTD.
QOSは、品質の審査担当者が第3部(モジュール3)の全体像を把握できるよう各項に関する十分な内容を含むものでなければならない。QOSは、申請に係る医薬品の重要な事項を明確にするとともに、例えば、データや資料を作成する際、既存のガイドラインによらないときは、その妥当性を説明するものでなければならない。QOSは、第3部(モジュール3)の各項資料と第4部(モジュール4)及び第5部(モジュール5)の関連資料を総合的に考え合わせた重要事項の考察(CTD-非臨床関連部(モジュール)記載の毒性試験に基づく不純物の定性等)を含むものでなければならない(他の部(モジュール)の資料を引用するときは、該当個所が相互に参照できるようにする)。
QOSは、表や図を除き、通常、本文が40ページを超えないこと。生物薬品及び複雑な製造工程を用いて製造される医薬品については、表や図を除き、通常、本文が80ページを超えないこと。QOS本文中の下線で示した図表、数値等については、第3部(モジュール3)記載のものをそのまま利用することができる。
The QOS should include sufficient information from each section to provide the Quality reviewer with an overview of Module 3. The QOS should also emphasise critical key parameters of the product and provide, for instance, justification in cases where guidelines were not followed. The QOS should include a discussion of key issues that integrates information from sections in the Quality Module and supporting information from other Modules (e.g. qualification of impurities via toxicological studies discussed under the CTDS module), including cross-referencing to volume and page number in other Modules.
This QOS normally should not exceed 40 pages of text, excluding tables and figures. For biotech products and products manufactured using more complex processes, the document could be longer but normally should not exceed 80 pages of text (excluding tables and figures).
The italicised text below indicates where tables, figures, or other items can be imported directly from Module 3.
緒言
緒言では、販売名、原薬の一般名(common nameを含む)、企業名、剤型、含量(力価)、投与経路及び申請に係る適応症を示す。
INTRODUCTION
The introduction should include proprietary name, non-proprietary name or common name of the drug substance, company name, dosage form(s), strength(s), route of administration, and proposed indication(s).
2.3.S 原薬
2.3.S.1 一般情報
3.2.S.1 記載の事項
2.3.S Drug Substance
2.3.S.1 General Information
Information from 3.2.S.1 should be included.
2.3.S.2 製造
3.2.S.2 記載の事項について以下の要素を勘案して記述する。
- 製造業者に関する記述
- 適正な品質の原薬を恒常的に製造するための製造工程(出発物質、重要工程、再加工についての記述等を含む)及びその管理に関する概略
- 3.2.S.2.2 に示す流れ図
- 3.2.S.2.3 記載の原薬製造に係る出発物質及び生物起源の原料の記述
- 重要工程の選択、プロセス・コントロール並びに規格値/判定基準と、その妥当性に関する考察。特に、3.2.S.2.4 記載の重要中間体については記述する。
- 3.2.S.2.5 記載のプロセス・バリデーション/プロセス評価の記述
- 3.2.S.2.6 記載の開発過程における製造方法の重大な変更に関する簡潔な要約及び製品の同等・同質性を評価するための検討結果に基づく結論。製造方法の変更後のロットを使用した非臨床試験や臨床試験結果については、申請資料の非臨床試験や臨床試験を記載した箇所との相互参照ができるようにする。関する概括資料(以下「QOS」という。)は、第3部の資料をその範囲及び構成に即して要約したものである。QOSには、第3部(モジュール3)及びCTDの他の部分に含まれていない事項、データ及び考察を加えてはならない。
2.3.S.2 Manufacture
Information from 3.2.S.2 should be included:
- Information on the manufacturer;
- A brief description of the manufacturing process (including, for example, reference to starting materials, critical steps, and reprocessing) and the controls that are intended to result in the routine and consistent production of material(s) of appropriate quality;
- A flow diagram, as provided in 3.2.S.2.2;
- A description of the Source and Starting Material and raw materials of biological origin used in the manufacture of the drug substance, as described in 3.2.S.2.3;
- A discussion of the selection and justification of critical manufacturing steps, process controls, and acceptance criteria. Highlight critical process intermediates, as described in 3.2.S.2.4;
- A description of process validation and/or evaluation, as described in 3.2.S.2.5.
- A brief summary of major manufacturing changes made throughout development and conclusions from the assessment used to evaluate product consistency, as described in 3.2.S.2.6. The QOS should also cross-refer to the non-clinical and clinical studies that used batches affected by these manufacturing changes, as provided in the CTD-S and CTD-E modules of the dossier.
2.3.S.3 特性
- 新規化学薬品:
-3.2.S.3.1 記載の構造及び異性体に関する資料の概略。
-原薬が光学活性体であるときは、非臨床試験や臨床試験に使われたものが立体異性体のどちらか一方か、又は立体異性体の混合物であるかを示すとともに、市販のための製剤に使用される原薬の立体異性体の情報を示す。
2.3.S.3 Characterisation
- For NCE:
– A summary of the interpretation of evidence of structure and isomerism, as described in 3.2.S.3.1, should be included.
– When a drug substance is chiral, it should be specified whether specific stereoisomers or a mixture of stereoisomers have been used in the nonclinical and clinical studies, and information should be given as to the stereoisomer of the drug substance that is to be used in the final product intended for marketing.
- 生物薬品:
-3.2.S.3.1 記載の目的物質及び目的物質関連物質の記述と、一般的性質、特性及び特性解析データ(一次及び高次構造、生物活性等)の要約を示す。
- For Biotech:
– A description of the desired product and product-related substances and a summary of general properties, characteristic features and characterisation data (for example, primary and higher order structure and biological activity), as described in 3.2.S.3.1, should be included.
- 新規化学薬品及び生物薬品:
-合成過程、製造過程あるいは変化・分解等によって生じる可能性がある不純物及び実際に検出された不純物に関するデータを要約するとともに、個別の不純物及び不純物全体について規格値/判定基準を設定するための根拠を要約する。非臨床試験・臨床試験に供されたロットと市販製造しようとする工程で製造された典型的なロットの不純物レベルについて比較考察する。不純物の設定限度値の妥当性について述べる。
-3.2.S.3.2 記載のデータの表形式のまとめ。適宜、図示。
- For NCE and Biotech:
– The QOS should summarise the data on potential and actual impurities arising from the synthesis, manufacture and/or degradation, and should summarise the basis for setting the acceptance criteria for individual and total impurities. The QOS should also summarise the impurity levels in batches of the drug substance used in the non-clinical studies, in the clinical trials, and in typical batches manufactured by the proposed commercial process. The QOS should state how the proposed impurity limits are qualified.
– A tabulated summary of the data provided in 3.2.S.3.2, with graphical representation, where appropriate should be included.
2.3.S.4 原薬の管理
- 規格及び試験方法の妥当性、試験方法及びその分析法バリデーションに関する簡潔な要約を示す。
- 3.2.S.4.1 記載の規格及び試験方法を示す。
- 3.2.S.4.4 記載のロット分析の表形式のまとめ。適宜、図示。-3.2.S.3.2 記載のデータの表形式のまとめ。適宜、図示。
2.3.S.4 Control of Drug Substance
- A brief summary of the justification of the specification(s), the analytical procedures, and validation should be included.
- Specification from 3.2.S.4.1 should be provided.
- A tabulated summary of the batch analyses from 3.2.S.4.4, with graphical representation where appropriate, should be provided.
2.3.S.5 標準品又は標準物質
- 3.2.S.5 の記載内容について、適宜、表にして示す。
2.3.S.5 Reference Standards or Materials
- Information from 3.2.S.5 (tabulated presentation, where appropriate) should be included.
2.3.S.6 容器及び施栓系
- 3.2.S.6 の記載内容について、簡潔な記述と考察を行う。
2.3.S.6 Container Closure System
- A brief description and discussion of the information, from 3.2.S.6 should be included.
2.3.S.7 安定性
- 3.2.S.7.1 記載の実施した試験の要約(条件、ロット、試験方法)、その結果の簡潔な考察及び結論、適宜、貯蔵方法、リテスト期間又は有効期間について示す。
- 3.2.S.7.2 記載の承認後の安定性試験計画を示す。
- 3.2.S.7.3 記載の安定性試験結果の表形式のまとめ。適宜、図示。
2.3.S.7 Stability
- This section should include a summary of the studies undertaken (conditions, batches, analytical procedures) and a brief discussion of the results and conclusions, the proposed storage conditions, retest date or shelf-life, where relevant, as described in 3.2.S.7.1.
- The post-approval stability protocol, as described in 3.2.S.7.2, should be included.
- A tabulated summary of the stability results from 3.2.S.7.3, with graphical representation where appropriate, should be provided.
2.3.P 製剤
2.3.P.1 製剤及び処方
3.2.P.1 の記載内容を示す。
3.2.P.1 記載の処方を示す。
2.3.P Drug Product
2.3.P.1 Description and Composition of the Drug
Information from 3.2.P.1 should be provided.
Composition from 3.2.P.1 should be provided.
2.3.P.2 製剤開発の経緯
3.2.P.2 記載内容及びデータに関する考察を示す。
必要に応じ、臨床試験に用いられた各処方の組成及びその溶出特性の一覧表を示す。
2.3.P.2 Pharmaceutical Development
A discussion of the information and data from 3.2.P.2 should be presented.
A tabulated summary of the composition of the formulations used in clinical trials and a presentation of dissolution profiles should be provided, where relevant.
2.3.P.3 製造
- 3.2.P.3 記載の以下の事項を示す。
- 製造業者に関する記述
- 適正な品質の製剤を恒常的に製造するための製造工程及びその管理に関する概略
- 3.2.P.3.3 に示す流れ図。
- 3.2.P.3.5 記載のプロセス・バリデーション/プロセス評価の簡潔な記述
2.3.P.3 Manufacture
Information from 3.2.P.3 should include:
- Information on the manufacturer.
- A brief description of the manufacturing process and the controls that are intended to result in the routine and consistent production of product of appropriate quality.
- A flow diagram, as provided under 3.2.P.3.3.
- A brief description of the process validation and/or evaluation, as described in 3.2.P.3.5.
2.3.P.4 添加剤の管理
3.2.P.4 記載の添加剤の品質に関する簡潔な要約を示す。
2.3.P.4 Control of Excipients
A brief summary on the quality of excipients, as described in 3.2.P.4, should be included.
2.3.P.5 製剤の管理
規格及び試験方法の妥当性についての簡潔な要約、試験方法、その分析法バリデーション及び不純物の特性の要約を示す。
3.2.P.5.1 記載の規格及び試験方法を示す。
3.2.P.5.4 記載のロット分析の表形式のまとめ。適宜、図示。
2.3.P.5 Control of Drug Product
A brief summary of the justification of the specification(s), a summary of the analytical procedures and validation, and characterisation of impurities should be provided.
Specification(s) from 3.2.P.5.1 should be provided.
A tabulated summary of the batch analyses provided under 3.2.P.5.4, with graphical representation where appropriate should be included.
The post-approval stability protocol, as described in 3.2.P.8.2, should be provided.
2.3.P.6 標準品及び標準物質
3.2.P.6 の記載事項について、適宜、表にして示す。
2.3.P.6 Reference Standards or Materials
Information from 3.2.P.6 (tabulated presentation, where appropriate) should be included.
2.3.P.7 容器及び施栓系
3.2.P.7 の記載内容について、簡潔な記述と考察を行う。
2.3.P.7 Container Closure System
A brief description and discussion of the information in 3.2.P.7 should be included.
2.3.P.8 安定性
実施した試験の要約(条件、ロット、試験方法)、試験結果及びデータ解析についての簡潔な考察並びに結論を示す。貯蔵方法、有効期間、適宜、使用時の保存条件及び有効期間に関する結論を示す。
3.2.P.8.3 記載の安定性試験結果の表形式のまとめ。適宜、図示。
3.2.P.8.2 記載の承認後の安定性試験計画を示す。
2.3.P.8 Stability
A summary of the studies undertaken (conditions, batches, analytical procedures) and a brief discussion of the results and conclusions of the stability studies and analysis of data should be included. Conclusions with respect to storage conditions and shelf-life and, if applicable, in-use storage conditions and shelf-life should be given.
A tabulated summary of the stability results from 3.2.P.8.3, with graphical representation where appropriate, should be included.
The post-approval stability protocol, as described in 3.2.P.8.2, should be provided.
2.3.A その他
2.3.A.1 製造施設及び設備
生物薬品:
3.2.A.1 に記載された施設に関する情報の要約を示す。
2.3.A Appendices
2.3.A.1 Facilities and Equipment
Biotech:
A summary of facility information described under 3.2.A.1 should be included.
2.3.A.2 外来性感染性物質の安全性評価
医薬品製造過程における内在性及び外来性感染性物質をコントロールする方策に関する考察を示す。
3.2.A.2 記載のウイルスクリアランス指数を一覧表として示す。
2.3.A.2 Adventitious Agents Safety Evaluation
A discussion on measures implemented to control endogenous and adventitious agents in production should be included.
A tabulated summary of the reduction factors for viral clearance from 3.2.A.2, should be provided.
2.3.A.3 新規添加剤
2.3.A.3 Excipients
2.3.R 各極の要求資料
必要に応じて、3.2.R 項に記載されている各極に特有な要求事項に対応する資料について、簡潔に記載する。
2.3.R Regional Information
A brief description of the information specific for the region, as provided under “3.2.R” should be included, where appropriate.





